Org. Synth. 2014, 91, 260-272
DOI: 10.15227/orgsyn.091.0260
Ni-catalyzed Reductive Cleavage of Methyl 3-Methoxy-2-Naphthoate
Submitted by Josep Cornella,
1 Cayetana Zarate,
1 and Ruben Martin
*1
Checked by Hande Gunduz, Richard P. Loach, and Mohammad Movassaghi
1. Procedure
A. Methyl 3-methoxy-2-naphthoate (1). An oven-dried 500 mL round-bottomed flask containing 3-hydroxy-2-naphthoic acid (10.1 g, 53.4 mmol, 1 equiv) (Note 1) and K2CO3 (29.5 g, 213.6 mmol, 4.0 equiv) (Note 2) is equipped with an oval magnetic stir bar (34 mm x 12 mm), argon inlet and a rubber septum. The flask is evacuated and back-filled with argon (this sequence is repeated three times over a period of 5 min) and the argon atmosphere is maintained throughout the reaction using an argon manifold system. The flask is charged through the septum via syringe with anhydrous DMF (120 mL) (Note 3), and iodomethane (17.3 mL, 277.7 mmol, 5.2 equiv) (Note 4) is added dropwise over a period of 2 min. The flask is then immersed in a pre-heated oil bath (Note 5) at 40 ºC and the mixture is stirred for 14 h. After this time, the flask is allowed to reach room temperature. The septum is removed and saturated NH4Cl solution (250 mL) is added (Note 6). The mixture is transferred to a 500 mL separatory funnel and extracted with EtOAc (5 x 50 mL) (Note 7). The combined organic layers are dried over anhydrous MgSO4 (30 g) (Note 8), filtered, rinsed with additional 30 mL of EtOAc and concentrated by rotary evaporation (from 760 mmHg to 36 mmHg, 42 ºC). The residue is purified by column chromatography on silica gel (Note 9), obtaining the title compound (1) as a colorless oil (11.1 g, 96%) (Notes 10 and 11).
B. Methyl 2-naphthoate (2). An oven-dried 100 mL pressure tube equipped with a Teflon screw-cap and a magnetic stir bar (13 mm x 5 mm) is brought into a nitrogen-filled glovebox, charged with methyl 3-methoxy-2-naphthoate (1) (5.13 g, 23.7 mmol, 1 equiv), bis(cyclooctadiene)nickel(0) (Ni(COD)2) (0.33 g, 1.18 mmol, 0.05 equiv) (Note 12), tricyclohexylphosphine (PCy3) (0.66 g, 2.37 mmol, 0.10 equiv) (Note 13) and toluene (47.5 mL) (Note 14). After addition of 1,1,3,3-tetramethyldisiloxane (TMDSO) (4.20 mL, 23.8 mmol, 1.0 equiv) (Note 15), the pressure tube is closed with the Teflon screw-cap, brought back to the fume hood and immersed in a pre-heated oil bath at 110 ºC. After 17 h, the resulting black solution is allowed to cool to room temperature, is filtered through a plug of Celite® (36 g) (Note 16) in a filter funnel (60 mm diameter) and is eluted with 130 mL of EtOAc (Note 17). The filtrate is concentrated by rotary evaporation (from 760 mmHg to 45 mmHg) and purified by column chromatography on silica gel (Note 18), obtaining the title compound as a white solid (3.9 g, 89% yield) (Notes 19 and 20).
2. Notes
1.
3-Hydroxy-2-naphthoic acid (98%) was purchased form Aldrich and used as received.
2.
Potassium carbonate (>99%) was purchased from Sigma-Aldrich and used as received without any further purification.
3.
DMF anhydrous (content in H
2O < 10 ppm) was dried from an Instrument Solvent Purification System (J. C. Meyer Solvent Systems).
4.
Iodomethane (
MeI, >99%) was purchased from Sigma-Aldrich and kept in the fridge at 4 ºC. It was used as received.
5. Oil Bath: silicone oil ò=0.97, was purchased from VWR and used as received (working temperature from -40 ºC to +200 ºC).
6.
NH4Cl was purchased from VWR. The solution was prepared using 30 g of
NH4Cl in 900 mL of distilled water.
7.
Ethyl acetate was purchased from Sigma-Aldrich and used as received.
8. Anhydrous
magnesium sulfate was purchased from VWR and used as received.
9. Column chromatography was performed on 170 g of silica gel
(230-400 mesh SiliaFlash®P60), purchased from Silicycle. It was wet packed in a 5 cm diameter column using hexanes/ethyl acetate (95/5) and the crude material was loaded into the column by means of a Pasteur pipette (the remaining residue was loaded in the minimal amount of hexanes/ethyl acetate (95/5)). Fractions of 40 mL were collected at 0.5 mL/s rate, eluting with hexanes/ethyl acetate (95/5). All fractions containing the desired product were combined, concentrated by rotary evaporation (from 760 mmHg to 38 mmHg) and dried overnight at 1.1 mmHg.
10. A second reaction on identical scale also provided 11.1 g (96%) of pure product.
11. Compound
1 has the following physical properties: R
f 0.27 (hexanes/ethyl acetate (90:10));
1H NMR
pdf(400 MHz, CDCl
3) δ: 3.97 (s, 3 H), 4.02 (s, 3 H), 7.22 (s, 1 H), 7.39 (ddd,
J = 8.1, 6.9, 1.2 Hz, 1 H), 7. 3 (ddd,
J = 8.2, 6.9, 1.2 Hz, 1 H), 7.75 (dd,
J = 8.3, 0.7 Hz, 1 H), 7.83 (dd,
J = 8.3, 0.7 Hz, 1 H), 8.32 (s, 1 H);
13C NMR
pdf(100 MHz, CDCl
3) δ: 51.87, 55.56, 106.46, 121.45, 124.07, 126.15, 127.19, 128.07, 128.33, 132.40, 135.78, 155.37, 166.34. IR (neat, cm
-1): 1732, 1632, 1281, 1210, 1074. HRMS Calcd for C
13H
12O
3: 217.0859, Found 217.0844. Anal Calcd for C
13H
12O
3: C, 72.21; H, 5.59; found: C, 71.95; H, 5.55. The submitters report that the product was isolated as a white solid with mp = 48-50 °C. Checkers determined the purity of the isolated compound
1 by GC-MS analysis (Column: Agilent Technologies, HP-5ms, 30 m x 0.250 mm, 0.25 mm. Max temp 350 °C; GC equipment: Agilent Technologies, 7890 A - Model number: G1033A; Detector: Agilent Technologies 5975C inert MSD with triple axis detector - Model number: G3171A). GC Temperature Program: Initial 50 ºC 1 min, Ramp 40 ºC/min up to 250 ºC. Hold for 3 min. Total run time: 9 min. Front Inlet (SS Inlet) flow: 19.00 mL/min Column flow: 1.000 mL/min. Retention time: 6.834 min.
Submitters also determined the purity of the isolated compound
1 by GC analysis (Column: Agilent Technologies, HP-5ms, 30 m x 0.250 mm, 0.25 mm, 19091S-433. Max temp 325 ºC; GC equipment: Agilent Technologies, 7890 A - Model number: G3440A; Detector: Agilent Technologies 5975C inert MSD with triple axis detector - Model number: G3171A). GC Temperature Program: Initial 50 ºC for 1 min, Ramp 40 ºC/min up to 250 ºC. Hold for 3 min. Total run time: 9 min. Column flow: 1.00 mL/min. Retention time: 7.099 min.
12.
Bis(cyclooctadiene)nickel(0) (
Ni(COD)2, >98%) was purchased from Strem Chemicals and stored at -35 ºC in the glove-box. It was used without further purification.
13.
Tricyclohexylphosphine (
PCy3, 97%) was purchased from Strem Chemicals and kept in the glovebox. It was used as received.
14. Toluene anhydrous (content in H
2O < 10 ppm) was dried from an Instrument Solvent Purification System (J. C. Meyer Solvent Systems).
15.
1,1,3,3-Tetramethyldisiloxane (97%) was purchased from Aldrich and used as received.
16. Celite®545 coarse was purchased from EMD Chemicals and used as received.
17. A filter plate (90 mm x 65 mm) was loaded with 36 g of Celite® and packed with 50 mL
EtOAc. Extra 130 mL of
EtOAc were used to elute the reaction mixture. The filtrate was collected in a 1 L round-bottomed flask.
18. Column chromatography was performed on 80 g of silica gel (230-400 mesh SiliaFlash®P60), purchased from Silicycle. It was wet packed in a 5 cm diameter column using hexanes/ethyl acetate (99/1) and the crude material was loaded into the column by means of a Pasteur pipette. (The remaining residue was loaded in the minimal amount of hexanes/ethyl acetate (99/1)). Fractions of 12 mL were collected eluting with 1 L hexanes/ethyl acetate (99/1), 0.5 L of hexanes/ethyl acetate (97/3) and finally 0.5 L hexanes/ethyl acetate (95/5). All fractions containing the desired product were combined, concentrated by rotary evaporation (from 760 mmHg to 45 mmHg) and dried for 30 min at 0.03 mmHg.
19. A second run on similar scale provided 4.5 g (82%) of pure product.
20. Compound
2 has the following physical properties: R
f 0.27 (hexanes/ethyl acetate (99:1)) mp = 73.7-74.5 ºC;
1H NMR
pdf(400 MHz, CDCl
3) δ: 3.99 (s, 3 H), 7.52-7.63 (m, 2 H), 7.87 (dt,
J = 8.1, 0.7 Hz, 2 H), 7.95 (dt,
J = 8.6, 0.7 Hz, 1 H), 8.08 (dd,
J = 8.6, 1.7 Hz, 1 H), 8.63 (s, 1 H);
13C NMR
pdf(100 MHz, CDCl
3) δ: 52.12, 125.15, 126.55, 127.33, 127.68, 128.07, 128.14, 129.27, 130.99, 132.42, 135.44, 167.15; IR (neat, cm
-1): 1711, 1233, 1201, 1154, 1130, 1100; HRMS [M + H] Calcd for C
12H
10O
2: 187.0759, Found 187.0750; Anal. calcd for C
12H
10O
2: C, 77.40; H, 5.41; found: C, 76.83; H, 5.36. Checkers determined the purity of the isolated compound
2 by GC-MS analysis (Column: Agilent Technologies, HP-5ms, 30 m x 0.250 mm, 0.25 mm. Max temp 350 ºC; GC equipment: Agilent Technologies, 7890 A - Model number: G1033A; Detector: Agilent Technologies 5975C inert MSD with triple axis detector - Model number: G3171A). GC Temperature Program: Initial 50 ºC 1 min, Ramp 40 ºC/min up to 250 ºC. Hold for 3 min. Total run time: 9 min. Front Inlet (SS Inlet) flow: 19.00 mL/min Column flow: 1.00 mL/min. Retention time: 6.073 min. Submitters also determined the purity of the isolated compound
2 by GC-MS analysis (Column: Agilent Technologies, HP-5ms, 30 m x 0.250 mm, 0.25 μm, 19091S-433. Max temp 325 ºC; GC equipment: Agilent Technologies, 7890 A - Model number: G3440A; Detector: Agilent Technologies 5975C inert MSD with triple axis detector - Model number: G3171A). GC Temperature Program: Initial 50 ºC for 1 min, Ramp 40 ºC/min up to 250 ºC. Hold for 3 min. Total run time: 9 min. Column flow: 1.000 mL/min. Retention time: 6.316 min.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
TThese procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Aryl methyl ethers are highly ubiquitous in nature. The importance of such motifs in organic synthesis is primarily associated to their unique role as directing groups, thus allowing a wide number of powerful synthetic transformations (Scheme 1, path a). Among these, particularly interesting are the
ortho-metalation;
2 electrophilic aromatic substitution;
3 Friedel-Crafts-type reactions;
4 ortho5, meta6 or
para7 C-H bond-functionalization reactions; etc. Importantly, all these transformations occur with an exquisite chemo- and regioselectivity control. In sharp contrast, unbiased and unactivated arenes are known to promote similar types of reactions, but in most instances harsh reaction conditions are required, thus making these transformations not particularly attractive from a synthetic standpoint due to regio- and chemoselectivity issues (Scheme 1, path b).
8 In recent years, elegant approaches based upon C-H bond-functionalization reactions have overcome most of these drawbacks. Despite the advances realized, an
ortho-directing group is typically required, and these procedures are not yet attractive enough as the cleavage of such groups still constitutes a tremendous challenge.
9
Despite formidable advances in the cross-coupling arena, this field of expertise is largely dominated by the use of organic halides as coupling counterparts. To such end, chemists have been challenged to come up with more flexible strategies from easily available precursors that do not require the use of halide waste. Recently, metal-catalyzed C-O bond-cleavage has become a powerful alternative to commonly employed organic halides.
10 Among their advantages is that such C-O electrophiles can be derived from simple and cheap phenols, thus enhancing the flexibility in catalytic design. Among the different C-O electrophiles, the utilization of simple aryl methyl ethers constitutes the best alternative because aryl methyl ethers are the simplest derivatives from all phenol series. Unfortunately, however, the high activation energy of the C-OMe bond constitutes a serious drawback to be overcome. Recently, we developed a new strategy based upon a metal-catalyzed reductive cleavage of C-OMe bonds as a means to use aryl methyl ethers as temporary removable directing groups (Scheme 1, path c).
11, 12 In this manner, such a technique represents a powerful alternative for arene functionalization that is difficult to accomplish using common synthetic organic techniques.
The Ni-catalyzed reductive cleavage was distinguished by its wide scope, including challenging substrate combinations. The protocol turned out to be widely tolerant of functional groups as silyl groups, esters, amides, acetals, amines and heterocycles remained intact (Scheme 2). Not surprisingly, extended π-systems were much more reactive than anisole derivatives, a common requisite in many C-O bond cleavage reactions. Although tentative, we postulate that such observation is in line with the known ability of extended π-systems to bind the Ni center in a η
2-fashion via the Dewar-Chatt-Duncanson model, thus retaining, unlike a regular arene, certain aromaticity that provides an extra stabilization. Interestingly, we found that the presence of an electron-withdrawing directing group in an
ortho-position allowed for the cleavage of C-OMe bonds in anisole derivatives.
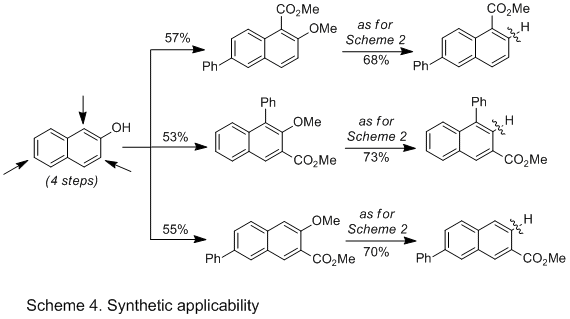
Our protocol could also be amenable for site-selectivity, as C-OMe bonds could be reductively cleaved in the presence of other C-OMe bonds, likely due to subtle steric as well as electronic differences (Scheme 3). Importantly, selectivity was also observed in highly complex molecules such as estradiol or quinine-type derivatives, thus reinforcing the notion that these transformations can potentially be used as a manifold for natural product diversity (Scheme 3). While one might question the synthetic applicability of a reductive cleavage event, we demonstrated that our technique might be useful for building up molecular complexity using the aryl methyl ether as a temporary protecting group. Indeed, we found that 2-naphthol could be easily transformed into three different regioisomeric naphthalenes by using the known ability of C-O bonds to direct functionalization at different positions on the arene followed by our Ni-catalyzed reductive cleavage event. These results can hardly be underestimated; indeed, it would be rather difficult to imagine a synthetic technique capable of rapidly accessing different regioisomers from a common precursor.
Most recently, we have shed light on the mechanism of this rather appealing transformation, by concluding that the commonly accepted Ni(0)/Ni(II) mechanism via oxidative addition into the C-OMe is not operating.
13 Indeed, we gathered evidence, both experimentally and computationally, that putative catalytically active Ni(I) intermediates come into play. Such species are likely generated via the comproportionation event of an initially formed Ni(II) species.
In conclusion, we have found a functional group tolerant Ni-catalyzed reductive cleavage of aryl methyl ethers that occurs with a wide substrate scope. Practicality aside, we believe such a reaction might open up new perspectives in the field of C-O bond-cleavage, a less-explored but vibrant area of expertise, and we expect that this method will find immediate application in organic synthesis.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Methyl 3-methoxy-2-naphthoate; (1) (13041-60-6)
3-Hydroxy-2-naphthoic acid; (92-70-6)
K2CO3: Potassium carbonate; (584-08-7)
MeI: Iodomethane; methyl iodide; (74-88-4)
Methyl 2-naphthoate; (2) (2459-25-8)
Ni(COD)2: Bis(cyclooctadiene)nickel(0); (1295-35-8)
PCy3: tricyclohexylphosphine; (2622-14-2)
TMDSO: 1,1,3,3-Tetramethyldisiloxane; (3277-26-7)

|
Ruben Martin received his PhD in 2003 from the University of Barcelona with Prof. Antoni Riera. In 2004, he moved to the Max-Planck Institut für Kohlenforschung as a Humboldt postdoctoral fellow with Prof. Alois Fürstner. In 2005 he then undertook further postdoctoral studies at MIT with Prof. Stephen L. Buchwald as a MEC-Fulbright fellow. In September 2008 he initiated his independent career as an assistant professor at the Institute of Chemical Research of Catalonia (ICIQ) and in July 2013 he was promoted to associate professor. His research is focused on the development of new metal-catalyzed activation of inert bonds. |

|
Dr. Josep Cornellà was born in Barcelona and graduated with distinction from the University of Barcelona (Spain) with a M.Sc in Organic Chemistry in 2008. Subsequently, he joined the group of Prof. Igor Larrosa at Queen Mary University of London (United Kingdom). In early 2012, he earned his PhD working on the development of metal-catalyzed decarboxylative transformations. In 2012, he was granted a Marie Curie Fellowship at Prof. Ruben Martin’s group at ICIQ and he is currently studying the discovery of novel transformations involving metal-catalyzed C-O bond activation and CO2 fixation. |

|
Cayetana Zárate received her B.Sc. from the University of Valladolid in 2012 with Extraordinary Award. During her B.Sc. she was an undergraduate fellow for two years carrying out research in gold-catalyzed reactions at Prof. Espinet´s group. In October 2012 she began graduate studies under the supervision of Prof. Ruben Martin at ICIQ, where she earned her M.Sc from the University Rovira i Virgili. She is currently pursuing her PhD studies at Prof. Ruben Martin´s group in the area of metal-catalyzed C-O bond-activation. |

|
Richard Loach was born in Birmingham (U.K.) and graduated from Imperial College, London in 2003 with a B.Sc in Chemistry. In 2007 he joined the research group of Professor John Boukouvalas at Laval University in Québec (Canada). In 2013, he earned his PhD for his studies on the total syntheses of novel γ-hydroxybutenolide natural products. In 2014, he was granted a FRQNT fellowship to pursue his postdoctoral research in Professor Mohammad Movassaghi’s group at MIT. He is currently working on the total synthesis of alkaloid natural products. |

|
Hande Gunduz was born in Istanbul (Turkey) and received her B.Sc. from Istanbul Technical University in 2009. In the same year she began her Ph.D. studies in the research group of Professor Naciye Talinli, focusing on the use of dioxinone chemistry for the synthesis of anticancer compounds. In 2013, she was granted a Fullbright Fellowship and joined Professor Mohammad Movassaghi’s research group at MIT as a visiting graduate student. She worked on the total synthesis of alkaloid natural products.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved