Checked by Brian M. Cochran and Margaret Faul
1. Procedure
A. (S)-N-(1-Hydroxy-3,3-dimethylbutan-2-yl)picolinamide (2). A 1 L one-necked round-bottomed flask equipped with a 3.0 cm x 1.4 cm, egg-shaped, Teflon-coated magnetic stirring bar is sealed with a septum and connected via needle adapter to a two-tap Schlenk adapter attached to an oil bubbler and a nitrogen/vacuum manifold (Note 1). The flask is dried with a heat gun under vacuum and cooled under a stream of nitrogen. The flask is charged with 2-picolinic acid (1) (6.15 g, 50.0 mmol, 1.00 equiv) (Note 2), evacuated and back-filled with nitrogen three times, then charged with dichloromethane (300 mL, 0.17 M) (Note 3) and N-methylmorpholine (7.59 g, 8.25 mL, 75.0 mmol, 1.50 equiv). The flask is cooled in an ice/water bath and iso-butylchloroformate (6.86 mL, 7.17 g, 52.5 mmol, 1.05 equiv) is added dropwise over 30 min by syringe pump. The reaction mixture is stirred for an additional 30 min while remaining submerged in the ice/water bath. A separate 100 mL one-necked round-bottomed flask is sealed with a septum and connected via needle adapter to the two-tap Schlenk adapter and manifold, dried with a heat gun under vacuum, and allowed to cool under a stream of nitrogen. This flask is charged with (S)-tert-leucinol (6.45 g, 55.0 mmol, 1.10 equiv), dichloromethane (40 mL), and N-methylmorpholine (6.07 mL, 5.56 g, 55.0 mmol, 1.10 equiv). The resulting clear solution is taken up in a syringe and transferred dropwise using a syringe pump over the course of 1 h to the stirring reaction mixture in the ice/water bath. The cooling bath is removed, and the pale gold colored reaction mixture is stirred for an additional 6 h at 23 °C. Upon consumption of starting material (Note 4), the mixture is quenched at ambient temperature with a single addition of an aqueous solution of saturated NH4Cl (50 mL), diluted with additional H2O (25 mL), and transferred into a 1 L separatory funnel. The phases are separated, and the aqueous phase is extracted with CH2Cl2 (3 x 100 mL). The combined organic phases are washed with an aqueous solution of saturated NaHCO3 (1 x 50 mL) and brine (1 x 50 mL). The combined organic phases are dried over Na2SO4 (10 g, 15 min while agitating), filtered through a M pore glass frit, and concentrated by rotary evaporation (28 °C, 15 mmHg). Excess N-methylmorpholine is further removed by placing the crude residue under high vacuum (< 12 mmHg, 12 h) to provide a pale red solid (Note 5). The crude residue is dissolved in 10 mL of acetone and purified via silica gel flash chromatography (Note 6). The combined product-containing fractions are concentrated by rotary evaporation (40 °C, 15 mmHg) to yield a solid, which is dried under high vacuum (< 12 mmHg, 12 h) to afford (S)-N-(1-hydroxy-3,3-dimethylbutan-2-yl)picolinamide (2) as a white amorphous solid (9.88-9.95 g, 44.4-44.8 mmol, 89-90% yield) (Note 7).
2. Notes
1. A two-tap Schlenk adapter connected to a bubbler and an argon/vacuum manifold is illustrated in Yu, J.; Truc, V.; Riebel, P.; Hierl, E.; Mudryk, B.
Org. Synth.
2008,
85, 64-71.
2.
Iso-butylchloroformate (98%) and
2-picolinic acid (99%) were purchased from Acros Organics and used without further purification.
N-Methylmorpholine (redistilled, >99.5%),
thionyl chloride (>99%),
sodium methoxide (95%),
ammonium hexafluorophosphate (99.9%), and
(S)-tert-leucinol (98%) were purchased from Sigma Aldrich and used as received. The submitters prepared
(S)-tert-leucinol as described by Krout, M. R.; Mohr, J. T.; Stoltz, B. M.
Org. Synth. 2009, 86, 181-193.
4-Chlorophenylboronic acid (98%) and
3-methylcyclohexen-1-one (98%) were purchased from Combi-Blocks and used as received. The submitters noted a variation in ee and yield with different commercial sources and batches of
4-chlorophenylboronic acid, presumably due to the presence of variable amounts of boroxine.
Palladium(II) trifluoroacetate (97%) was purchased from Strem Chemicals and
3-methylcyclohexen-1-one (98%) was purchased from Combi-Blocks, and each was used as received.
3.
Methylene chloride (> 99.8%, anhydrous),
toluene (99.8%, anhydrous), methanol (99.8%, anhydrous) and reagent grade
1,2-dichloroethane (99.8%, anhydrous) were purchased from Sigma Aldrich and used as received. In house deionized water was used without alteration.
4. The reaction can be monitored by TLC analysis using 3:2 hexanes/acetone as the eluent (E. Merck Silica gel 60 F254 precoated plates, 250 nm), visualizing with UV fluorescence quenching and
p-anisaldehyde staining or iodine staining. Product amide (
2) R
f = 0.42, impurity R
f = 0.38 (consistent with
3,3-dimethyl-2-(picolinamido)butyl picolinate),
N-methylmorpholine R
f = 0.06 (iodine stain).
Picolinic acid (
1) and
tert-leucinol remain at the baseline (R
f = 0), as determined by TLC.
5. If
N-methylmorpholine is not removed prior to chromatography, it will overload the silica gel column and will contaminate product fractions. Excess
N-methylmorpholine complicates product solidification post chromatography.
6. Silica gel column dimensions: 5 cm diameter x 20 cm height, ca. 220 g silica gel (RediSep RF column from Teledyne Isco, catalog 69-2203-422), eluting with 4:1 hexanes/acetone. Pre-run of 200 mL is collected before fractions are taken. Fractions are collected in 18 mm x 150 mm test tubes. Fraction purity can be assayed by TLC analysis using 3:2 hexanes/acetone with UV visualization. Product (
2) R
f = 0.42, impurity R
f = 0.38 (consistent with
3,3-dimethyl-2-(picolinamido)butyl picolinate),
N-methylmorpholine R
f = 0.06 (iodine stain). Both starting materials remain at the baseline (R
f = 0).
7.
(S)-N-(1-Hydroxy-3,3-dimethylbutan-2-yl)picolinamide (
2) exhibited the following characterization data: R
f = 0.42 with 3:2 hexanes/acetone; mp 78.7-79.8 °C;
1H NMR
pdf(400 MHz, CDCl
3) δ: 1.05 (s, 9 H), 2.60 (br s, 1 H), 3.70 (dd,
J = 8.4, 11.2 Hz, 1 H), 3.96-4.03 (m, 2 H), 7.44 (dd,
J = 5.2, 6.8 Hz, 1 H), 7.85 (dt,
J = 1.2, 8.0 Hz, 1 H), 8.19 (d,
J = 8.0 Hz, 1 H), 8.32 (br d,
J = 8.0 Hz, 1 H), 8.56 (d,
J = 4.8 Hz, 1 H);
13C NMR
pdf(100 MHz, CDCl
3) δ: 27.0, 33.7, 60.5, 63.6, 122.4, 126.2, 137.4, 148.0, 149.6, 165.5; IR (film): 3390, 3241, 2966, 1646, 1535, 1464, 1435, 1368, 1293, 1241, 1083, 1054, 1022, 999, 863, 750, 697 cm
-1; HRMS (Multimode ESI/FTMS)
m/z calcd for C
12H
19N
2O
2 [M + H]
+: 223.1441, found: 223.1439; [α]
D24 = -9.09 (
c 1.728, CHCl
3); Anal calcd for C
12H
18N
2O
2: C, 64.84; H, 8.17; N, 12.61. Found: C, 64.65; H, 8.19; N, 12.60.
8. The submitters used an oil bath heated to 60 °C.
9. Care should be taken to ensure the addition rate is initially very slow to avoid formation of a brown, tar-like byproduct. By addition of the first 3.5 mL over 30 min and the remaining solution (~ 23 mL) over 40 min by syringe pump, no byproduct was seen.
10.
(S)-N-(1-Chloro-3,3-dimethylbutan-2-yl)picolinamide hydrochloride salt exhibited the following characterization data:
1H NMR
pdf(400 MHz, DMSO-
d6) δ: 0.93 (s, 9 H), 3.88-3.97 (m, 2 H), 4.08 (dt,
J = 3.6, 9.6 Hz, 1 H), 7.68 (ddd,
J = 1.2, 4.8, 7.2 Hz, 1 H), 8.09 (dt,
J = 1.2, 7.6 Hz, 1 H), 8.15 (d,
J = 7.6 Hz, 1 H), 8.68-8.71 (m, 2 H), 13.24 (br s, 1 H);
13C NMR
pdf(100 MHz, DMSO-
d6) δ: 26.6, 35.3, 45.1, 59.4, 122.5, 126.9, 138.8, 147.9, 149.0, 163.7; IR (Neat): 3192, 3023, 2953, 2922, 1680, 1601, 1561, 1518, 1472, 1345, 1291, 1211, 1181, 1036, 1002, 971 cm
-1; HRMS (MultiMode ESI/FTMS)
m/z calcd for C
12H
18ClN
2O [M+H]
+: 241.1102, found 241.1103; [α]
22D = +25.09 (
c 1.132, MeOH). Anal calcd for C
12H
18Cl
2N
2O: C, 52.00; H, 6.55; N, 10.11; Cl 25.58. Found: C, 52.29; H, 6.59; N, 10.10; Cl, 25.57. A broad resonance at ~ 5 ppm in the
1H NMR spectrum is found with incomplete drying of the product; by D
2O quenching we determined this resonance to be the hydrate.
11.
Sodium methoxide is used from a freshly opened bottle or retrieved from storage in a nitrogen-atmosphere glovebox, free of adventitious moisture. The addition to the reaction mixture is exothermic.
12. The reaction can be monitored by TLC analysis using 3:2/hexanes:acetone as the eluent, visualizing with UV fluorescence quenching and
p-anisaldehyde staining. Product R
f = 0.44, starting material R
f = 0.32.
13.
Toluene is added to prevent the crude ligand from exposure to concentrated
sodium methoxide. The ligand is unstable to concentrated acids or bases.
14. After
toluene was added, approx 140-160 mL of solvent were removed on the rotovap. During the work-up and phase separation, a persistent rag and emulsions formed. The rag is to be kept with the top organic layer.
15. Silica gel column dimensions: 5 cm diameter x 20 cm height, ca. 200 g silica gel (Silica Gel ZEOprep® 60 ECO 40-63 Micron from American International Chemical, Inc. It is necessary to use this specific silica gel to suppress reversion to
(S)-N-(1-hydroxy-3,3-dimethylbutan-2-yl)picolinamide during chromatography, which was observed when employing silica gel from other commercial sources. The submitters report the reaction's yield is decreased by 5-10% when another silica gel is used.) (Note: The checker used this brand of Silica Gel as received from Prof. Stoltz's lab. No other silica brands were examined) eluting with 4:1 hexanes/acetone. Fractions are collected in 18 mm x 150 mm test tubes. Fraction purity can be assayed by TLC analysis using 3:2 hexanes/acetone with UV visualization. Product (
3) R
f = 0.44. This slow eluent is used for chromatography to remove a yellow impurity of similar polarity that contaminates the white solid product if the column is eluted with higher polarity eluent. This impurity appears to be an indiscrete decomposition product not readily identified. This impurity is not easily removed by recrystallization, and contaminated material should be resubmitted to flash chromatography.
16.
(S)-tert-ButylPyOx (
3) exhibited the following characterization data R
f = 0.44 with 3:2 hexanes/acetone; mp 70.4-71.0 ºC;
1H NMR
pdf(400 MHz, CDCl
3) δ: 0.99 (s, 9 H), 4.13 (dd,
J = 8.8, 10.4 Hz, 1 H), 4.32 (t,
J = 8.4 Hz, 1 H), 4.46 (t,
J = 8.8 Hz, 1 H), 7.39 (ddd,
J = 1.2, 4.4, 7.2 Hz, 1 H), 7.77 (dt,
J = 1.2, 8.0 Hz, 1 H), 8.10 (d,
J = 8.0 Hz, 1 H), 8.72 (d,
J = 4.0 Hz, 1 H);
13C NMR
pdf(100 MHz, CDCl
3) δ: 26.0, 34.0, 69.3, 76.5, 124.0, 125.4, 136.5, 147.0, 149.7, 162.5; IR (film): 2954, 2903, 2867, 1640, 1565, 1466, 1358, 1345, 1272, 1244, 1211, 1095, 1037, 967 cm
-1; HRMS (MultiMode ESI/APCI)
m/z calcd for C
12H
17N
2O [M+H]
+: 205.1335, found 205.1336; [α]
22D = -91.85 (c 5.09, CHCl
3); Anal calcd for C
12H
16N
2O, C 70.56, H 7.90, N 13.71, found C 71.09, H 8.12, N 13.29. Purity of
3 was assessed at 99.3 wt% by quantitative
1H NMR in CDCl
3 using methyl phenyl sulfone as a standard. If desired, the white solid can be recrystallized from minimal hot
n-heptane, with crystals collected at -20 °C (after cooling to room temperature) as white needles. However, the ligand is more difficult to weigh in this state due to its tendency to cling with static electricity. Enantiomeric excess can be determined via analytical chiral SFC (Jasco SFC utilizing a Chiralcel OB-H column (4.6 mm x 25 cm) obtained from Daicel Chemical Industries, Ltd) with visualization at 210 nm and flow rate of 5 mL/min eluting with 10% MeOH/CO
2. Major enantiomer retention time: 2.51 min, minor enantiomer retention time:
2.20 min. (In addition to the high optical purity as seen by polarimetry, the material was analysed by SFC using the method above and the ligand was determined to be 99.7% ee.) Attempts were made to separate enantiomers via analytical chiral HPLC using various columns (Chiralcel OD-H, Chiralcel OJ-H, Chiralpak AD, Chiralpak AS, Chiralcel OB-H) and solvent systems, however adequate separation of peaks was not achieved. In the absence of an analytical chiral SFC, optical rotation measurements can be utilized to give the enantiomeric excess by way of optical purity calculations (optical purity (%) =
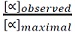
x 100). The optical rotation listed above may be used as α
maximal and a sufficiently large concentration (e.g. c 5.00) as well as multiple trials should be used in order to minimize error. It is imperative to store the ligand in a desiccator to minimize hydrolysis, which results in diminished enantiocontrol in the conjugate addition reaction. For long-term storage, the ligand should be stored frozen under an inert atmosphere; we recommend a nitrogen atmosphere glovebox freezer.
17. Vigorous stirring that results in a visible vortex is essential for reaction conversion, as dispersion of water throughout the
1,2-dichloroethane is necessary. After water is added to the reaction, a viscous gel is formed which prevented the magnetic stir bar from spinning. The reaction was manually agitated until the stir bar became free.
18. The reaction progress can be monitored by TLC analysis with 4:1 hexanes/ethyl acetate, using a
p-anisaldehyde stain; product (
6) R
f = 0.38 (stains yellow/orange with fresh
p-anisaldehyde stain),
3-methyl-2-cyclohexenone (
4) R
f = 0.19 (stains tan/brown), 4,4'-dichloro-1,1'-biphenyl R
f = 0.65 (does not stain). Reaction times typically are 12-24 h, with conversion slowing considerably after the first several hours.
19. Silica gel plug dimensions: 3 cm diameter x 4 cm height, ca. 12 g silica gel.
20. The product is very crystalline. When loading onto the column, the product may crystallize. Silica gel column dimensions: 5 cm diameter x 20 cm height, ca. 220 g silica gel (RediSep RF column from Teledyne Isco, catalog 69-2203-422). The column is eluted with 92:8 hexanes/ethyl acetate until the product is collected. Fractions are collected in 18 mm x 150 mm test tubes. Fraction purity can be assayed by TLC analysis using 4:1 hexanes/ethyl acetate with UV fluorescence quenching visualization and heating with
p-anisaldehyde; product (
6) R
f = 0.38, (stains yellow/orange with fresh
p-anisaldehyde stain),
3-methyl-2-cyclohexenone (
4) R
f = 0.19
(stains tan/brown), 4,4'-dichloro-1,1'-biphenyl R
f = 0.65 (does not stain).
21.
(R)-3-(4-Chlorophenyl)-3-methylcyclohexanone (
6) exhibited the following characterization data R
f = 0.38 (hexanes/ethyl acetate 4:1),
1H NMR
pdf(400 MHz, CDCl
3) δ: 1.30 (s, 3 H), 1.60-1.69 (m, 1 H), 1.85-1.94 (m, 2 H), 2.12-2.19 (m, 1 H), 2.31 (t,
J = 6.8 Hz, 2 H), 2.42 (d,
J = 14.4 Hz, 1 H), 2.83 (d,
J = 14.4 Hz, 1 H), 7.23-7.25 (m, 2 H), 7.27-7.30 (m, 2 H);
13C NMR
pdf(100 MHz, CDCl
3) δ: 21.9, 29.9, 37.9, 40.7, 42.6, 52.9, 127.1, 128.6, 132.0, 145.8, 210.9; IR (thin film): 2944, 2865, 1693, 1589, 1492, 1450, 1432, 1352, 1265, 1229, 1092, 1010, 952, 824 759, 737, 721 cm
-1; HRMS (MultiMode ESI/APCI)
m/z calcd for C
13H
14ClO [M+H]
+: 223.0884, found 223.0885; [α]
D22 = -65.8 (
c 1.912, CDCl
3); Anal calcd C
13H
15ClO: C 70.11, H 6.79, O 7.18, Cl 15.92 found C 69.73, H 7.02, O 7.49, Cl 15.88. Enantiomeric excess of 93% is determined via analytical chiral HPLC (Agilent 1100 Series HPLC utilizing a Chiralcel OB-H column (4.6 mm x 25 cm) obtained from Daicel Chemical Industries, Ltd) with visualization at 254 nm and flow rate of 1 mL/min eluting with 1%
iso-propanol/hexanes. The sample was prepared in a 1:1 v/v solution of
iso-propanol/hexanes. Major enantiomer retention time 16.47 min, minor enantiomer retention time
14.55 min.
3. Discussion
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved