Org. Synth. 2016, 93, 50-62
DOI: 10.15227/orgsyn.093.0050
Nickel-Catalyzed Synthesis of Ketones from Alkyl Halides and Acid Chlorides: Preparation of Ethyl 4-Oxododecanoate
Submitted by Alexander C. Wotal, Donald C. Batesky, and Daniel J. Weix
*1
Checked by Yongda Zhang and Chris H. Senanayake
1. Procedure
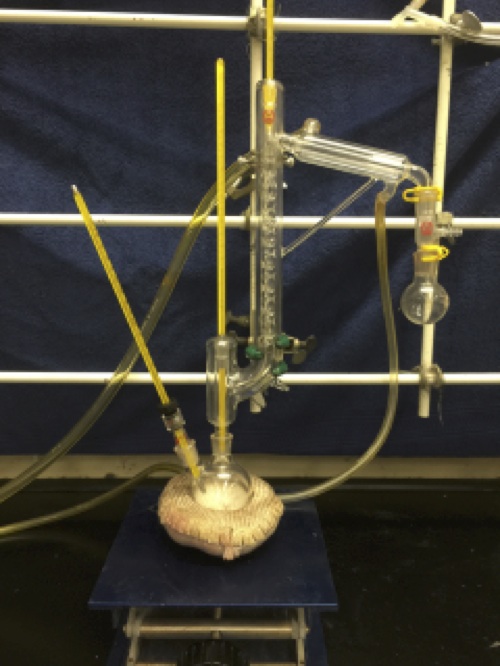
A four-necked 500-mL Jacket reactor (
Note 1) with 24/40 ground glass joints is equipped with a septum bearing a thermocouple connected to a J kem temperature controller, an addition funnel with a septum, and a nitrogen inlet connected to a Firestone valve. The center neck is equipped for mechanical stirring (
Figure 1) (
Note 2). The reactor is connected to a chiller (
Note 3). The reactor is charged with
nickel(II) chloride ethylene glycol dimethyl ether complex (1.11 g, 4.90 mmol, 0.050 equiv) (
Note 4),
4,4'-di-tert-butyl-2,2'-dipyridyl (1.48 g, 5.39 mmol, 0.055 equiv) (
Note 5) and
manganese powder (16.68 g, 303.6 mmol, 3.1 equiv) (
Note 6). The system is evacuated and refilled with
nitrogen three times. Anhydrous
N,N-dimethylacetamide (
Note 7) (200 mL) is added to the reactor through the additional funnel by syringe. The reaction mixture is stirred for 1 h under nitrogen flow. During this time the green reaction mixture turns to a dark green/black suspension. The stirred mixture is then cooled to an internal temperature at 0-2 °C over 0.5 h.
1-Iodooctane (24.0 g, 98.0 mmol, 1.0 equiv) and
ethyl succinyl chloride (25.45 g, 146.9 mmol, 1.5 equiv) (
Note 8) are successively charged into the reactor via the addition funnel by syringe. The color of the reaction mixture changed to brown in 5 min. The reaction mixture is vigorously stirred at 0 to 2 °C for 14 h and then is allowed to warm to room temperature over 0.5 h and then stirred for another 3-6 h at room temperature (
Note 9).
Figure 1. Apparatus Assembly
The stirred reaction mixture is diluted by adding 1:1 hexanes/methyl tert-butyl ether (200 mL), resulting in the formation of a dark green/black precipitate. The mixture is drained from the reactor (Notes 10 and 11). The solids are removed by filtration through a celite bed (Note 12). The black solids are scraped off the top of the celite and transferred to a 500-mL Erlenmeyer flask and stirred with 250 mL of deionized water for 1-2 min. The insoluble manganese salts are removed by vacuum filtration through the same celite bed and rinsed with deionized water (20 mL). The water filtrate is combined with the organic filtrate from the reaction mixture. The resulting mixture is transferred to a 1-L separatory funnel. The aqueous layer is extracted with 1:1 hexanes/methyl tert-butyl ether (200 mL). The combined organic layers are washed with deionized water (200 mL) followed by a final wash with saturated sodium chloride solution (250 mL). The product solution mixture is transferred to a 1-L Erlenmeyer flask and stirred over 20 g of anhydrous magnesium sulfate for 15-20 min. The magnesium sulfate is removed by vacuum filtration of the dried solution using a 7 cm Büchner funnel. The solvents are distilled from the crude product mixture using a rotary evaporator (30 °C, 25-30 mmHg).
A very small forerun (ca. 3 mL) is removed by distillation at 150 °C under vacuum (5-6 mmHg) (
Figure 2). The pot is cooled to 23 °C before the vacuum is released, after which the forerun is discarded (
Note 11). The brownish crude product is then loaded onto a column pre-prepared with 50 g silica gel in hexanes (
Note 13). The product is eluted with 10% MTBE in hexanes (500 mL). All the eluted solution is collected. After the solvent is removed by rotary evaporation (50 °C, 50 mmHg),
ethyl 4-oxododecanoate (19.90 g, 84%) is collected as a pale yellow oil (Notes
14,
15, and
16).
Figure 2. Distillation Assembly
2. Notes
1. The reactor was washed with ~100 mL of concentrated
hydrochloric acid followed by 200-300 mL deionized water.
Methanol (200 mL) was added to the reactor and then heated to reflux for 2-3 h. After the reactor was cooled to room temperature, MeOH was drained from the reactor and the reactor dried with the jacket temperature of 100 °C under vacuum (150-200 mmHg) for 1-2 h. The reactor was cooled to 23 °C under vacuum and then refilled with
nitrogen before use.
2. The stirrer paddle must not be in contact with the bottom or sides of the flask. The stirrer paddle was generally held ~10 mm above the bottom of the flask to prevent mechanical activation of the reducing agent, which could cause lower product yields (see Figure 1). The submitters use a mechanical stirring (glass bearing, glass stirrer rod, 6 cm PTFE paddle, air driven motor). Minimal amounts of Dow Corning high vacuum grease were used to seal all ground glass joints, and heavy mineral oil (mineral oil white, heavy, Mallinckrodt) was used to lubricate the glass stir rod inside the 24/40 standard taper adapter.
3. The model of the chiller is Julabo FS18. The submitters used ice-water in a 10-L container to cool the reaction mixture.
4.
Nickel(II) chloride ethylene glycol dimethyl ether complex (dme or glyme) (min. 97%) was obtained from Strem and used as received.
5.
4,4'-tert-Butyl-2,2′-dipyridyl (98%) was obtained from Aldrich and used as received.
6.
Manganese powder (-325 mesh, ≥99% trace metals basis) was obtained from Aldrich and used as received. Reactions run with a smaller amount of
manganese (2 equiv) still proceeded to completion, but took longer to reach completion. Due to the low cost of Mn metal, an excess was used for convenience.
7. Anhydrous
N,N-dimethylacetamide (99.8%) was obtained from Aldrich and used as received. The submitters used
N,N-dimethylacetamide (99%) from Alfa, degassed with argon, and dried using cartridge no. VAC 104458 in a VAC Solvent Purifier (Vacuum Atmospheres Co., Hawthorne, CA 90250, USA).
8. Commercial
1-iodooctane (98%, Aldrich) was obtained from Aldrich and used as received. The submitters filtered the commercial
1-iodooctane through a pad of basic alumina to remove any acidity and copper stabilizer.
Ethyl succinyl chloride (95%) was obtained from Aldrich and was used as received. The checker observed mild exotherm during the addition. The internal temperature increased from 1.7 °C to 3.3 °C with the jacket temperature at 0 °C.
9. In two small pilot runs, a sample of the reaction mixture was checked for the presence of
1-iodooctane by gas chromatography, but in these pilot runs, only a trace was seen in the forerun and no monitoring was done on the large-scale runs. The checker monitored the reaction by GC-MS, using an Agilent HP-5MS colum (30 m x 0.25 mm x 0.25 µm). Helium flow rate 1 mL/min; Initial temp: 50 °C (2 min), ramp 25°C/min to 200 °C (hold for 2 min). Retention times are
1-iodooctane (5.47 min) and
ethyl 4-oxododecanoate (8.05 min). Only trace amount of
1-iodooctane was observed once the reaction mixture was warmed to room temperature.
10. These solids include remaining
manganese, water soluble salts and some product. The filtration is carried out using a 7-cm Büchner funnel with a Celite® bed on a Whatman GF/A glass microfiber filter set on a 1-L filtration flask. The checkers observed slow filtration without the Celite® bed. The Celite® (C212-5W Celite®, 545 filter aid) was 1 cm thick and obtained from Fisher Scientific and used as received.
11. If the reactor is contaminated with residual metal, conc.
HCl followed by water rinse can be used to clean the reactor. These rinses are for the purpose of cleaning the glass and are not added to the filtrate.
12. Instead of purification by chromatography, the submitters isolated the product by distillation at reduced pressure. A 50-mL, 2-necked round-bottomed flask is equipped with a thermometer on the side 14/20 joint and a 20-cm vacuum jacketed Vigreux column with a built-in water cooled side arm, a vacuum adapter, and 25-mL receiving flask (
Figure 3). The apparatus is placed under aspirator vacuum (~18-20 mmHg). A very small forerun of about 2-3 mL is distilled, boiling up to 90 °C. After the last forerun is removed, the pot is allowed to cool to 40 °C before the vacuum released. The forerun is discarded and the 20-cm Vigreux column is exchanged for a shorter 6-cm Vigreux column/head with a vacuum side arm adapter attached to a high-vacuum line and a clean 25-mL receiving flask, cooled in ice water. The hold up using the longer Vigreux column is avoided by switching to the shorter column, hence avoiding overheating and possible degradation of the product (
Figure 4) The product is distilled at a pot temperature ranging from 120-140 °C, with a head temperature of 101-102 °C (1 mmHg). For this operation, a high-vacuum pump instead of a water aspirator was used.
|

|
|
Figure 3. Distillation Assembly Figure
|
4. Purification by Distillation
|
|
13. Silica gel (technical grade, pores size 60, 230-400 mesh particle size) was received from Aldrich and was used as received. The length of the silica gel is about 10 cm in the column, while the column for flash chromatography has 50 mm ID and 45.7 cm effective length with a sealed coarse porosity fritted disc.
14. The product exhibited the following physical properties: yellow liquid.
1H NMR
pdf(400 MHz, CDCl
3) δ: 0.88 (t,
J = 6.5 Hz, 3H), 1.20-1.23 (m, 13H), 1.59 (t,
J = 7.0 Hz, 2H), 2.44 (t,
J = 7.5 Hz, 2H), 2.57 (t,
J = 6.7 Hz, 2H), 2.71 (t,
J = 6.5 Hz, 2H), 4.14 (q,
J = 7.2 Hz, 2H).
13C NMR
pdf(100 MHz, CDCl
3) δ: 14.0, 14.1, 22.6, 23.8, 28.0, 29.1, 29.2, 29.3, 31.8, 37.0, 42.8, 60.5, 172.8, 209.1. FTIR (neat, cm
-1): 1734 (C=O of ester), 1715 (C=O of ketone), 1181 (C-O). HRMS: Calcd for C
14H
27O
3 (M+H
+): 243.1960; found: 243.1968.
15. Purity was assessed at 98 wt% by quantitative
1H NMR using dimethyl fumarate as an internal standard.
16. The yield of a second duplicate run was 20.0 g (84%).
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Because of the prevalence of alkyl alkyl ketones in natural products, their use in biological probes,
2 and their versatility as intermediates in organic synthesis,
3 a wide variety of methods have been developed to access them. The most commonly used methods are the addition of a reactive organometallic reagent to an aldehyde followed by oxidation and the addition of pre-formed organometallic nucleophiles to carboxylic acid derivatives.
2 While both of these protocols are effective, the organometallic reagents can be limiting with complex, functionalized molecules. More recently, cross-coupling methods based upon the less reactive alkyltin,
4 alkylzinc,
5,6 dialkylzinc,
7 or alkylboron
8 reagents have been developed.
We recently reported a different strategy, the nickel-catalyzed coupling of carboxylic acid chlorides or thioesters with alkyl iodides under reducing conditions (Table 1).
9 This work built upon previous studies of Mukaiyama
10 and electrochemical work from Nédélec, Périchon, Amatore, and Jutand.
11 Relatively sterically hindered ketones could be made by this protocol, a challenge for other coupling methods (Table 1), and the reaction conditions tolerated a variety of functional groups, including protected amines, amide N-H bonds, a vinyl boronic ester, protected alcohols, and protected alkynes (
Tables 1 and 2).
These conditions have proven to be general in our hands and in the hands of others. For example, Gong reported modified conditions that enable coupling with anhydrides
12 as well as the use of tertiary alkyl halides,
13 aryl acid chlorides,
14 and methyl
p-toluenesulfonate ester.
15 It is also important to note that one-pot protocols for the generation and coupling of acid chlorides
14 and anhyrides
13 have been reported. In addition, by substituting a chiral bis(oxazoline) ligand for bipyridine, Reisman was able to realize the enantioconvergent coupling of a secondary benzyl chloride to form enantioenriched α-aryl ketones.
16 Finally, although several mechanisms had been proposed in the literature for these reactions by ourselves and others, recent investigations have led to a revised mechanism. We proposed a connection between the cross-electrophile coupling of aryl halides with alkyl halides based upon mechanistic studies of isolated acylnickel intermediates
17 and subsequent work by Gong
12 suggested that the mechanism is nearly identical to the one we proposed for the coupling of aryl halides with alkyl halides.
18Although these studies have demonstrated that cross-electrophile ketone synthesis has promising functional group compatibility and scope, no reports on a preparative scale have appeared. Because these reactions are heterogeneous, we anticipated that stirring method, stirring rate, and activity of the metal powder could all play a role in success. In this case, as in a previous report on a related reaction,
19 avoiding mechanical activation of the metal powder by keeping the stirrer paddle off the bottom of the flask and ensuring turbulent mixing (as with a Morton flask) were the key differences from the smaller-scale procedure. In our previous report,
19 low activity of the zinc powder sometimes led to long induction periods, but no such variability was observed with
manganese powder and the reactions proceed even at 0 °C. This difference may be due to
in situ activation of the metal powder by
HCl, formed by reaction of the acid chloride with adventitious water.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Nickel (II) chloride ethylene glycol diethylether: Nickel (II) chlorideglyme, Nickel(II) chloride dimethoxyethane complex; (29046-78-4)
4, 4', Di-tert-butyl-2,2'-bipyridine; 4-tert-Butyl-2-(4-tert-butyl-2- pyridyl)pyridine, (72914-19-3)
N,N-Dimethylacetamide; (127-19-5)
Manganese; (7439-96-5)
1-Iodooctane; (629-27-6)
Ethyl succinyl chloride: Ethyl 4-chloro-4-oxobutyrate; (14794-31-1)

|
Donald C. Batesky graduated from Purdue University with a B.S. in Chemistry in 1954. Following 38 years with Eastman Kodak Co. as a new product development chemist and in management, Don worked 15 years with Aldrich Chemical Co. developing new products. In 2014, Don began a third career at University of Rochester in the Weix group. In his career spanning over 50 years, he has prepared almost 4000 organic chemicals. |

|
Daniel J. Weix was raised in Oak Creek, WI. After undergraduate training with Tom Katz at Columbia University, graduate work with Jonathan Ellman at Berkeley, and postdoctoral training with John Hartwig at Yale and Illinois, he started his independent career at Rochester in 2008, where he is currently an associate professor. His research program in the area of transition metal catalysis has been focused on developing the concepts of cross-electrophile coupling. Cross-electrophile coupling has proven to be a useful approach to synthesis both in academia and in industry. |

|
Alexander C. Wotal was raised in Palatine, Illinois. In 2006, he began studying chemistry at William Rainey Harper College, Palatine, Illinois under Prof. Daniel J. Stanford. He completed his B.S. in 2009 under Prof. Jeffrey B. Johnson at Hope College, Holland, Michigan. Alexander then joined the group of Daniel J. Weix at University of Rochester, Rochester, New York where he studied Ni-catalyzed ketone-forming cross-electrophile coupling reactions and their mechanisms. In 2014, he participated in a 1-year cooperative position at GlaxoSmithKline pharmaceutical company under Dr. Jared Spletstoser and Dr. Qi Jin developing antibacterial agents. |

|
Yongda Zhang was born in Zhejiang province, China. He received his B.A. degree in chemistry from Hangzhou University in 1994. He earned his Ph.D. in organic chemistry at Fudan University under the guidance of Prof. Dawei Ma (Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences) and Prof. Fenggang Tao (Fudan University) in 2000. After postdoctoral research with Prof. Alan J. Kennan and additional postdoctoral training with Prof. Tomislav Rovis at Colorado State University, he joined Boehringer Ingelheim Pharmaceuticals Inc. in Ridgefield, CT, where currently he is Principal Scientist. His research interests include design and development of innovative and practical processes for large-scale production of active pharmaceutical ingredients and synthetic methodology. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved