Org. Synth. 2016, 93, 228-244
DOI: 10.15227/orgsyn.093.0228
Oxyboration: Synthesis of Borylated Benzofurans
Submitted by Darius J. Faizi, Nicole A. Nava, Mohammad Al-Amin, and Suzanne A. Blum
*1
Checked by Nicholas A. Weires and Neil K. Garg
1. Procedure
Caution! Sodium hydride liberates hydrogen gas, which presents a fire risk. Care should be taken during handling and the chemical should be quenched with caution.
A.
2-(Phenylethynyl)phenol (
3). A flame-dried 1-L round-bottomed flask equipped with a stir bar (
Note 1) is charged with
2-iodophenol (
1, 14.0 g, 63.6 mmol, 1.00 equiv), copper iodide (1.09 g, 5.71 mmol, 0.09 equiv), and
bis(triphenylphosphine)palladium dichloride (1.33 g, 1.89 mmol, 0.03 equiv) (Notes
2 and
3). The flask is then covered with a rubber septum. A needle is then placed in the septum and the flask is then flushed with nitrogen (
Note 4). The flask is then kept under a dynamic nitrogen atmosphere.
Toluene (320 mL) is then added via cannula, followed by
diisopropylamine (8.90 mL, 63.6 mmol, 1.00 equiv) using a 24-mL syringe in one portion (Notes
5 and
6).
Phenylacetylene (
2) (10.5 mL, 95.4 mmol, 1.50 equiv) is then added via a 24-mL syringe over five min (Notes
7 and
8, Figure 1). The reaction is stirred for 8.5 h at 25 °C (
Note 9).
Figure 1. Step A - Color before and after addition of 2
The septum is removed, and the reaction mixture is filtered through a plug of celite (Note 10). The mixture is then transferred to a 1-L separatory funnel. The filter flask is rinsed with 100 mL of ethyl acetate and added to the separatory funnel. The organic layer is rinsed with saturated aqueous ammonium chloride (1 × 50 mL), saturated aqueous brine (1 × 50 mL), and then transferred to a 1-L Erlenmeyer flask containing anhydrous sodium sulfate (55 g) and allowed to sit for 15 min. The solid sodium sulfate is then removed by filtration through a medium porosity 150-mL filter funnel (200 mmHg applied vacuum), and the resulting solution is collected into a 1-L round-bottomed flask and concentrated by rotary evaporation (35 °C, 10 mmHg) (Note 11). The dark brown oil is then purified by flash chromatography using an eluent mixture of 12% ethyl acetate in hexanes as the eluent (Note 12). The collected fractions are placed in a 250-mL round-bottomed flask and then concentrated by rotary evaporation (35 °C, 10 mmHg) and then placed under high vacuum (25 °C, 0.2 mmHg) for 13 h (Note 13). The brown residue is recrystallized from hexanes to afford 6.34 g (51.4%) of 2-(phenylethynyl)phenol (3) as a brown crystalline solid (Notes 14, 15, 16, and 17).
B.
4,4,5,5-tetramethyl-2-(2-phenylbenzofuran-3-yl)-1,3,2-dioxaborolane (4). This procedure is carried out in an argon-filled glove box (
Note 18). A 500-mL round-bottomed flask is charged with a stir bar,
sodium hydride (0.81 g, 31 mmol, 1.0 equiv, 92% wt.), and
toluene (192 mL) (Notes
19 and
20). In a separate 20-mL vial, compound
3 (6.1 g, 31 mmol, 1.0 equiv) is dissolved in
toluene (15 mL). This solution is drawn into a 24-mL syringe with a long-stem needle. The vial is then rinsed with
toluene (3 mL), which is also drawn into the same syringe. This solution is then added to the reaction flask over 30 min (
Note 21, Figure 2). The reaction is then stirred for an additional 15 min.
Figure 2. Step B - Part a - Reaction mixture during addition of 3
In a separate 20-mL vial,
B-chlorocatecholborane (4.8 g, 31 mmol, 1.0 equiv) is dissolved in
toluene (15 mL) (
Note 22). This solution is then drawn up into another 24-mL syringe equipped with a long stem needle. The vial is rinsed with
toluene (3 mL), which is then drawn up into the same syringe. This combined solution is then added to the reaction mixture in the round-bottomed flask over 1 min and then the flask is capped with a glass stopper equipped with a Teflon ring. The reaction mixture is then stirred for 30 min (
Note 23). In another 20‑mL vial,
1,3-bis(2,6-diisopropylphenyl-imidazol-2-ylidene)gold(I) chloride, also known as
IPrAuCl, (0.58 g, 0.94 mmol, 3.0 mol%) is dissolved in
toluene (18 mL) (
Note 24). This suspension is then transferred via pipette to the reaction mixture over 1 min. The vial is then rinsed with 10 mL
toluene and this rinse is added via pipette to the reaction mixture.
Sodium trifluoroacetate (1.3 g, 9.4 mmol, 0.30 equiv) is added to a 20-mL vial, and then added in one portion to the reaction mixture (
Note 25). This vial is then rinsed with
toluene (6 mL), and this
toluene is then added to the reaction mixture via pipette. The round‑bottomed flask is then capped with a glass stopper equipped with a Teflon ring, and a Keck plastic joint clip. The flask is placed in a pre-heated sand bath (60 °C) and allowed to stir in the glovebox for 21 h (Figure 3)
Figure 3. Step B - Part c - Reaction mixture heated to 60 °C
At this time, TLC indicated that the reaction is complete (Note 26). The mixture is then cooled to room temperature before triphenylphosphine (0.41 g, 1.6 mmol, 5.0 mol%) is added in one portion (Note 27). The reaction flask is then allowed to stir for 3 h. In a separate 100-mL round-bottomed flask, pinacol (11 g, 94 mmol, 3.0 equiv) is dissolved in triethylamine (65 mL, 470 mmol, 15 equiv) (Note 28). This solution is added to the reaction mixture via pipette over 4 min (Note 29). The flask is capped with a glass stopper, and the reaction mixture is stirred for 1 h. The flask is then taken out of the glovebox and the resulting suspension is filtered through celite, using toluene (200 mL) as a rinse (Note 30). The recovered liquid is transferred to a clean 1-L round-bottomed flask and concentrated by rotary evaporation (40 °C, 10 mmHg). The dark brown oil is then dissolved in dichloromethane (20 mL) and transferred to a 100-mL round-bottomed flask and then concentrated by rotary evaporation (30 °C, 10 mmHg). The mixture is then placed under high vacuum (0.2 mmHg) for 16 h (Note 31). Column chromatography using 20% dichloromethane in hexanes yielded 7.26 g (73%) of a light yellow solid after drying under high vacuum for 18 h (25 °C, 0.2 mmHg) (Notes 32, 33, and 34).
2. Notes
1. A cylindrical-shaped PTFE stir bar with dimensions of 3.7 cm x 0.9 cm x 1.0 cm was used.
2. The submitters sourced
2-iodophenol (98%) from two different suppliers. The submitters and checkers purchased
2-iodophenol (98%) from Acros and used it as received. Another batch was purchased by the submitters from Frontier Scientific, which was contaminated with a large amount of iodine. To remove the iodine, 10 g of the
2-iodophenol was dissolved in 300 mL of
ethyl acetate and placed in a 500-mL separatory funnel. The organic layer was then washed with saturated aqueous sodium thiosulfate (3 × 50 mL), brine (1 × 50 mL), and then dried over
sodium sulfate (55 g). The solution was then concentrated using rotary evaporation (40 °C, 10 mmHg) and then placed under high vacuum for 24 h (25 °C, 0.2 mmHg). The light yellow oil was then placed in the freezer for 1 h, and the resulting light yellow crystals were used in the reaction.
3.
Bis(triphenylphosphine)palladium dichloride (98%) was purchased from Sigma Aldrich and used as received. Copper iodide (98%) was purchased from Strem Chemicals Inc., and used as received.
4. The needle used was a BD PrecisionGlide® disposable needle, 20 gauge, short bevel, purchased from VWR International.
5.
Toluene (99.8%, HPLC grade) was purchased from Fisher Scientific and passed through a basic alumina push-still system before use.
Diisopropylamine (99.95%) was purchased from Sigma Aldrich and sparged with nitrogen for 15 min before use.
6. After addition of the
diisopropylamine, the solution turned from yellow to a red-orange color.
7.
Phenylacetylene (98%) was purchased from Alfa Aesar and sparged with nitrogen for 15 min before use.
8. The checkers added
phenylacetylene manually using a 20-gauge needle approximately 6 inches in length. The submitters utilized a syringe pump for this addition. After about 1 min, the solution changed from a red-orange to dark brown/black color.
9. The reaction was monitored via thin-layer chromatography on glass-backed silica gel TLC plates with a UV254 indicator, purchased from Merck Millipore International.
2-(Phenylethynyl)phenol, R
f = 0.42 (10%
ethyl acetate in hexanes).
10. The filtration was conducted as follows: a medium porosity 350-mL filter funnel with a 9.0 cm I.D. was charged with 64.1 g of celite (Celite 545 was purchased from EMD Millipore and used as received). The solid was then pre-wet with 200 mL of
toluene and then connected to a 1-L filter flask. The reaction mixture was then poured over the wet celite, and pressure was applied to pull the solution through the pad (200 mmHg). The celite pad was then rinsed with 200 mL of
toluene, and filtered using applied pressure (200 mmHg).
11. During this step, the submitters decided to transfer the solution to a 250-mL round-bottomed flask to aid in the subsequent chromatography step.
12. The column chromatography was run as follows: A flash column with an outer diameter of 11 cm and capacity of 2-L (Pyrex No. 1280) was charged with silica (Davisil 35-70 micron silica purchased from Grace Davidson; used as received) using a wet-pack method (1.0 kg of silica in 1950 mL of hexanes). This gave a silica bed of 9 inches in height. The crude mixture was then dissolved in 50 mL of
dichloromethane, and 15.0 g of silica was then added to the 250-mL round-bottomed flask containing the crude mixture. This mixture was then slowly concentrated using a rotary evaporator (25-35 °C, 50-10 mmHg), yielding a light brown powder. This powder was then dry loaded into the column [Alternatively, the crude mixture could be liquid loaded using a pipette]. Sand was then added to fill 2 inches above the silica. The column was then loaded with the eluent mixture (88:12 hexanes: ethyl acetate). The flow rate was approximately 66 mL/min, and 2500 mL of the eluent were allowed to drain before 300-mL fractions were collected. Fractions 1-13 were then collected as the product fractions; fractions 14-20 were collected as co‑elute.
13. The submitters found that trace
ethyl acetate would rapidly dissolve the material, resulting in issues during the recrystallization. Placing the compound under high vacuum (25 °C, 0.2 mmHg) for 9 h would remove all of the
ethyl acetate and make the recrystallization more effective.
14. The residue obtained from fractions 1-13 were recrystallized as follows: a 100-mL round-bottomed flask was charged with the brown solid, which was then dissolved in boiling hexanes (50 mL), capped with a ground glass stopper, and then allowed to cool to room temperature over 20 h. At this time, the round-bottomed flask was placed in an ice bath for 2 h, and then placed in the freezer (-20 °C) for an additional 2 h. The supernatant is filtered off at reduced pressure (200 mmHg) in a Buchner funnel (9 cm in diameter) with filter paper and the resulting brown solid crystals were washed with chilled hexanes (-20 °C, 1 × 5 mL) and then dried in a 250‑mL round-bottomed flask under high vacuum (25 °C, 0.2 mmHg) for 6 h. The first crop of crystals weighed 2.51 g. The filtrate was then concentrated by rotary evaporation (25 °C, 10 mmHg) and then concentrated under high vacuum (25 °C, 0.2 mmHg) for 2 h. The recovered brown solid was then dissolved in boiling hexanes (20 mL), capped with a ground glass stopper, and then allowed to cool to room temperature over 20 h. The cooling and filtration process was repeated as above, and the resulting crystals weighed 1.83 g, with a combined final recovered mass of 4.34 g.
15. The residue obtained from fractions 14-20 (the co-eluted fractions) were recrystallized as follows: a 100-mL round-bottomed flask was charged with the brown solid, which was then dissolved in boiling hexanes (40 mL), capped with a ground glass stopper, and then allowed to cool to room temperature over 20 h. At this time, the round-bottomed flask was placed in an ice bath for 2 h, and then placed in the freezer (-20 °C) for an additional 2 h. The supernatant is filtered off at reduced pressure (200 mmHg) in a Buchner funnel (9 cm in diameter) with filter paper and the resulting brown solid crystals were washed with chilled hexanes (-20 C, 1 × 5 mL) and then dried in a 250-mL round-bottomed flask under high vacuum (25 °C, 0.2 mmHg) for 6 h. This crop of crystals weighed 2.00 g.
16. The physical properties of
2-(phenylethynyl)phenol follow: R
f = 0.42 (10%
ethyl acetate in hexanes, visualized by short-wave UV);
1H NMR
pdf(CDCl
3, 500 MHz) δ: 5.83 (s, 1H), 6.92 (td,
J = 7.6, 1.0 Hz, 1H), 6.99 (dd,
J = 8.2, 0.7 Hz, 1H), 7.25-7.30 (m, 1H), 7.36-7.40 (m, 3H), 7.43 (dd,
J = 7.7, 1.5 Hz, 1H), 7.52-7.58 (m, 2H).
13C NMR
pdf(CDCl
3, 125 MHz) δ: 83.1, 96.5, 109.7, 114.9, 120.6, 122.5, 128.7, 129.0, 130.6, 131.76, 131.80, 156.6. IR (film): 3441, 3049, 1596, 1572, 1496, 750, 688 cm
-1. Melting Point: 69.5-70.6 °C. HRMS calc for C
14H
11O [M+H]
+: 195.0810. Found 195.0801. The purity of the compound was determined using qNMR: 14.2 mg of the product are dissolved in 0.8 mL of CDCl
3. Mesitylene, 11.0 mg (98+%, purchased from Alfa Aesar and used as received), is added.
1H NMR (500 MHz, CDCl
3) gave a product purity of 97.8%.
17. A second run on the same scale provided 6.49 g (53%) of the product.
18. This reaction is performed in a nitrogen-filled glove box because the boronic ester intermediate that is formed is highly sensitive to moisture (see Table 1 in the discussion section). The submitters were able to synthesize the desired borylated benzofuran using air-free techniques with septa-capped glassware and dynamic nitrogen at the bench instead of a glovebox, however the yield was substantially lower (50% vs 70% at the 0.86 mmol scale). All glassware used in the glove box was oven-dried overnight before being brought into the glove box.
19. A cylindrical-shaped PTFE stir bar with dimensions of 3.7 cm x 0.9 cm x 1.0 cm was used.
20.
Sodium hydride (95 wt%) was purchased from Sigma Aldrich. This material was stored in the nitrogen-filled glovebox, and was titrated to 92% using a spectroscopy-based titration method developed by Hoye.
2 Toluene (99.8%, HPLC grade) was purchased from Fisher Scientific and passed through a basic alumina push-still system, then brought into the glovebox by the checkers using a 500-mL Straus flask. The submitters utilized a push-still system that fed directly into the glovebox. It is important that the
toluene is water-free for this reaction.
21. The checkers performed this addition manually. The submitters utilized a syringe pump for this addition. A slight exotherm was noted. Care should be used during this step, as this reaction liberates hydrogen gas. Careful monitoring and very slow addition are of paramount importance. It is highly recommended to unplug any electronic devices/ionizers that are not in use for this reaction while the highly flammable hydrogen gas is being liberated.
22. The submitters used
B-chlorocatecholborane (98+%) purchased from TCI America. The checkers used
B-chlorocatecholborane (97%) purchased from Sigma Aldrich. This solid was stored in the glove box freezer, and subsequently allowed to warm to room temperature before use.
23. It is important to cover the reaction flask at this point, as the newly formed boronic ester is highly moisture sensitive.
24.
1,3-Bis(2,6-diisopropylphenyl-imidazol-2-ylidene)gold(I) chloride (99%) was purchased from Frontier Scientific and used as received.
25.
Sodium trifluoroacetate (98%) was purchased from Sigma Aldrich and was dried as follows: A 50-mL round-bottomed flask was charged with ca. 5 g of
sodium trifluoroacetate and a stir bar. The solid was dried under high vacuum (0.2 mmHg) at 110 °C for 18 h. The flask was then placed directly into the glovebox while remaining under vacuum.
26. The TLC was conducted using an eluent of 10% methylene chloride in hexanes. 4,4,5,5-Tetramethyl-2-(2-phenylbenzofuran-3-yl)-1,3,2-dioxa-borolane: R
f = 0.29 (10% DCM in hexanes)
27.
Triphenylphosphine (98+%) was purchased from Strem Chemicals and used as received.
28.
Pinacol (99%) was purchased from Alfa Aesar and used as received.
Triethylamine (99.5%+) was purchased from Sigma Aldrich and passed through a basic alumina push-still system before use. A large excess of
triethylamine was used so that it could fully solubilize the
pinacol (thus acting as both the base and the solvent)
29. A slight evolution of gas was noted.
30. The filtration was conducted as follows: a medium porosity 350-mL filter funnel with a 9.0 cm I.D. was charged with 64.1 g of celite (Celite 545 was purchased from EMD Millipore and used as received). The solid was then pre-wet with
toluene (200 mL) and then connected to a 500-mL filter flask. The reaction mixture was then poured over the wet celite, and pressure was applied to pull the solution through the pad (200 mmHg). The celite pad was then rinsed with
toluene (200 mL).
31. Although not necessary, placing the crude reaction mixture under high vacuum (0.2 mmHg) removes the excess
pinacol and
triethylamine, which reduces the total amount of material being placed on the column for purification. If the high vacuum step is omitted, it is advised to use a larger chromatographic column than the one described in this text.
32. The column chromatography was run as follows: A flash column with an outer diameter of 7 cm and capacity of 500-mL was charged with silica (Davisil 35-70 micron silica purchased from Grace Davidson; used as received.) using a wet-pack method (575 g of silica in 650 mL of hexanes). This gave a silica bed of 7 inches in length. The crude liquid mixture was then added to the top of the silica bed using a pipette; the reaction flask was rinsed with methylene chloride (3 × 5 mL) and then added to the silica. Sand was then added to fill 1.5 inches above the silica. The column was then loaded with the eluent mixture (80:20 hexanes: methylene chloride). The flow rate was approximately 53 mL/min, and 580 mL were then allowed to drain before fractions were collected in 50-mL test tubes (25 x 150 mm). Fractions 2-23 were collected as the product fractions. Fraction 1 was discarded, as it was contaminated with
triphenylphosphine.
33. The physical properties for
4,4,5,5-tetramethyl-2-(2-phenylbenzofuran-3-yl)-1,3,2-dioxaborolane follow: R
f = 0.29 (10% DCM in hexanes, visualized by short-wave UV).
1H NMR
pdf(CDCl
3, 500 MHz) δ: 1.41 (s, 12H), 7.23-7.31 (m, 2H), 7.38-7.48 (m, 3H), 7.49-7.53 (m, 1H), 7.99-8.03 (m, 1H), 8.14-8.20 (m, 2H);
11B NMR
pdf(CDCl
3, 96 MHz) δ: 30.8;
13C NMR
pdf(CDCl
3, 125 MHz) δ: 25.1, 83.8, 110.7, 123.1, 123.2, 124.4, 128.3, 129.2, 131.4, 133.4, 154.8, 163.1. IR (film): 3059, 2977, 1611, 1561, 1389, 1013, 747 cm
-1. Melting Point: 89.4-90.8 °C. HRMS calc. for C
20H
22BO
3 [M+H]
+: 321.1662. Found 321.1641. The purity of the compound was determined using qNMR: 28.4 mg of the product was dissolved in 0.8 mL of CDCl
3. 11.0 mg of mesitylene (98+%, purchased from Alfa Aesar and used as received) was added.
1H NMR (500 MHz, CDCl
3) gave a product purity of 97.4%.
34. The checkers also found that when Step B was performed on 15.5 mmol (3.05 g) scale with respect to
3, the yield of
4,4,5,5-tetramethyl-2-(2-phenylbenzofuran-3-yl)-1,3,2-dioxaborolane was 3.53 g (71%).
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Boron-element additions are a powerful class of synthetic reactions capable of furnishing boron-containing compounds that are adept at downstream functionalization. First described by Hurd
3 and developed by Brown,
4 hydroboration adds a B-H bond across unsaturated C-C bonds. This method of B-X bond addition has been extended to certain heteroatoms.
5 We recently reported the addition of B-O σ-bonds across alkynes.
6 Prior to this work, B-O s-bond addition across C-C unsaturated bonds was unknown, likely due to the strength of the B-O s bond, rendering transition metal-catalyzed oxidative addition, a common technique for borylation, difficult.
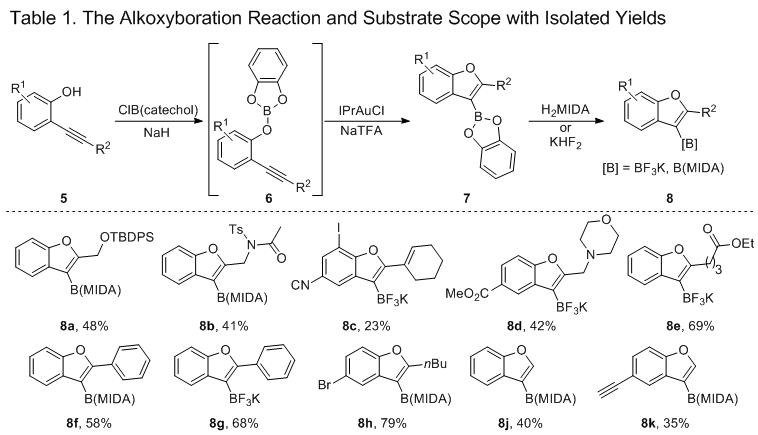
7 We hereby describe a robust procedure for the alkoxyboration of alkynes. This alkoxyboration method provides access to bench-stable borylated benzofurans isolated as the potassium organotrifluoroborate salt or the MIDA boronate ester.
8,9The outlined procedure results in the formation of 3-borylated benzofuran derivatives (Table 1). Beginning with 2-alkynylphenols (
5), the requisite boric ester intermediate (
6) is generated by deprotonation and electrophilic trapping by
B-chlorocatecholborane. Cyclization is effected by the catalyst IPrAuTFA, generated in situ from
IPrAuCl and NaTFA, producing the boronic ester intermediate (
7). For long-term stability, the catechol boronic ester was then transesterified to the more stable organotrifluoroborate salt or MIDA boronate ester (
8). A number of functional groups were tolerated by the alkoxyboration reaction, including siloxy (
8a), amide (
8b), nitrile (
8c), and ester (
8e) groups. The synthesized BF
3K salts
8 and MIDA esters
9 provide potential handles for further downstream functionalization resulting in the rapid synthesis of structurally complex compounds.
6
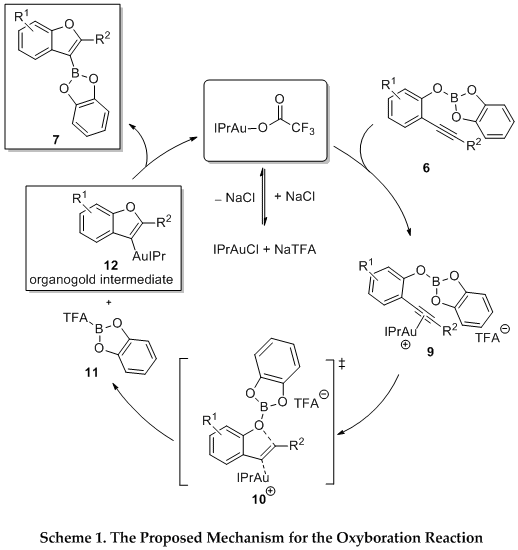
The mechanism of this alkoxyboration reaction is proposed to follow the pathway outlined in Scheme 1. Following the formation of the boronic ester intermediate, the catalyst IPrAuTFA could facilitate the generation of the
O-heterocyclic scaffold by coordinating to boric ester
6, yielding species
9, facilitating a nucleophilic attack that furnishes an organogold intermediate (
12) and boric ester
11. These two intermediates recombine to produce borylated alkoxyboration product (
7) and the catalyst is regenerated. Mechanistic experiments in our group have examined the role of the NaTFA counterion in the alkoxyboration reaction.
13
Since our first report of the alkoxyboration reaction, we have optimized several of the reaction parameters to enhance the ease of performing the reaction. Previously, generation of the requisite 2‑alkynylphenol starting materials,
5, required three steps: protection of the phenol group, Sonogashira coupling, and subsequent deprotection. Now using a procedure adapted from Tamaru, we synthesize
3 using a protecting-group free Sonogashira coupling reaction directly from
2-iodophenol (
1), thereby reducing the total number of synthetic steps to one in order to produce the desired 2-alkynylphenol
3, which is isolated in a reasonable yield.
10In addition to optimizing the synthesis of the starting 2-alkynylphenol substrates
3, we have examined the possibility of performing the key catalytic alkoxyboration reaction outside the glovebox for operational ease. In order to compare, both reactions were run on the 0.86 mmol scale, with the only variable being in or out of the glovebox. On this smaller scale, a lower yield was obtained for the in-box reaction compared to the large scale reaction reported in this manuscript. Although lower in isolated yield (50% vs. 70%), we have successfully run the oxyboration reaction outside of the glovebox using standard air-free techniques with septa-capped glassware. The lower yield for the out of the glovebox reaction is likely due to hydrolysis of the boric ester intermediate (
6). For the procedure described in this text, we decided to run the reaction in the glovebox in order to obtain the highest possible yield. Moreover, we have optimized the catalyst quenching (addition of PPh
3) to reduce the total reaction time. Finally, we expanded the transesterification step from MIDA esters and organotrifluoroborate salts to pinacol boronic esters using a procedure adapted from Ingleson.
11 The pinacol boronic esters are valuable because they are well-established cross-coupling partners and Michael addition partners in transition metal chemistry.
12In closing, an optimized oxyboration reaction, capable of furnishing 3-borylated benzofurans that are primed for downstream functionalization, is reported. The requisite starting materials are constructed in one step from commercial materials via a protecting-group-free Sonogashira reaction. The reaction conditions are mild and scalable.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
2-Iodophenol: Phenol, 2-iodo-; (533-58-4)
Copper iodide: Copper iodide; (1335-23-5)
Bis(triphenylphosphine)palladium dichloride: Palladium, dichlorobis(triphenylphosphine); (13965-03-2)
Toluene: benzene, methyl-; (108-88-3)
Diisopropylamine: ; 2-Propanamine, N-(1-methylethyl)-; (108-18-9)
Phenylacetylene: Benzene, ethynyl-; (536-74-3)
Sodium hydride: sodium hydride; (7646-69-7)
B-Chlorocatecholborane: 1,3,2-Benzodioxaborole, 2-chloro- (55718-76-8)
1,3-Bis(2,6-di-isopropylphenylimidazol-2-ylidene)gold(I)chloride: Gold, [1,3-bis[2,6-bis(1-methylethyl)phenyl]-1,3-dihydro-2H-imidazol-2-ylidene]chloro-; (852445-83-1)
Sodium trifluoroacetate: Acetic acid, 2,2,2-trifluoro-, sodium salt (1:1) (2923-18-4)
Triphenylphosphine: Phosphine, triphenyl- (603-35-0)
Pinacol: 2,3-Butanediol, 2,3-dimethyl- (76-09-5)
Triethylamine: Ethanamine, N,N-diethyl- (121-44-8)

|
Suzanne A. Blum received her B.S. from the University of Michigan in 2000, where she trained with Profs. Edwin Vedejs and Brian P. Coppola. She earned her Ph.D. degree at the University of California, Berkeley, studying the mechanisms of transition metal reactions, as an NSF Graduate Fellow under the mentorship of Profs. Jonathan A. Ellman and Robert G. Bergman. She completed an NIH Postdoctoral Fellowship with Prof. Christopher T. Walsh at the Harvard Medical School. In 2006, she began her independent career as an Assistant Professor at the University of California, Irvine. She was promoted to Associate Professor in 2012. |

|
Darius J. Faizi received his B.S. in chemistry from the University of California, Los Angeles in 2010, where he studied transition metal catalysis under the guidance of Professor Craig A. Merlic. He is currently pursuing his Ph.D. in organic chemistry under the guidance of Professor Suzanne A. Blum. His graduate research focuses on the transition-metal catalyzed addition of B-O bonds across C-C multiple bonds. |

|
Nicole A. Nava received her B.S. in chemistry from the University of California, Irvine in 2015. While there, she completed undergraduate research under the direction of Professor Suzanne A. Blum. She will begin pursuing a Ph.D. in organic chemistry at Boston College in Fall 2015. |

|
Mohammad Al-Amin was born in Chandpur, Bangladesh. He received his Ph.D. in synthetic organic chemistry in 2010 under the supervision of Professor Masahiro Yoshida and Professor Kozo Shishido at the University of Tokushima as a MEXT scholar. In 2010, he moved to Hokkaido University as a postdoctoral fellow under the direction of Professor Mitsuhiro Arisawa and Professor Satoshi Shuto. He is currently a postdoctoral fellow in the group of Professor Suzanne A. Blum at the University of California, Irvine. His main research interests are the development of new synthetic methods using transition metal catalysts. |

|
Nicholas A. Weires received his B.S. in chemistry from the University of Idaho in 2012, where he studied heterogeneous catalysis under the direction of Professor Jakob Magolan. He is currently pursuing his Ph.D. in organic chemistry in the laboratory of Professor Neil K. Garg at the University of California, Los Angeles. His graduate research has focused on the total synthesis of welwitindolinone alkaloids and the development of new C-C bond forming reactions of amides mediated by nickel catalysis. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved