1. Procedure (Note 1)
A.
Phosphinothioic amide, P,P-diphenyl (
2).
2,3 An oven-dried 300-mL three-necked round-bottomed flask equipped with a 2.5-cm Teflon-coated oval magnetic stirring bar, a small glass stopper, a reflux condenser topped with a 3-way glass stopcock fitted with an argon balloon, and a straight glass tube with a rubber septum is charged with
chlorodiphenylphosphine (
1) (10.0 g, 45.3 mmol, 1.0 equiv) and
sulfur (1.45 g, 45.3 mmol, 1.0 equiv). The flask is charged with dry
THF (75 mL) via a syringe and flushed with argon (Notes
2,
3, and
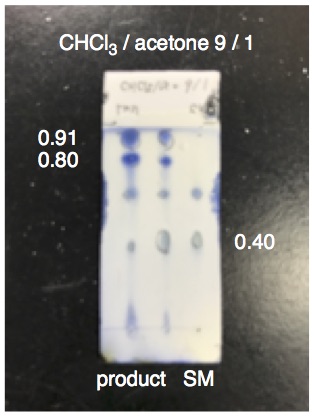
4). The reaction mixture is heated in an oil bath at 60 ºC (bath temperature) and stirred for 23 h under an argon atmosphere (Figure 1). The reaction mixture is a pale yellow homogeneous solution. The reaction progress is monitored by TLC analysis (Figure 2) (
Note 5) and
31P NMR (
Note 6). After consumption of
1, the straight glass tube is replaced with an
ammonia-gas line inlet, and the reflux condenser is replaced with a glass stopper. The small glass stopper is replaced with a pressure-resistant rubber hose attached to a 3-way glass stopcock to discharge the excess of
ammonia gas into a water bath. The mixture is cooled to 0 ºC in an ice bath, and the
ammonia gas, set to a constant gas-feed at a pressure of approx. 0.015 MPa is bubbled into the reaction mixture through a glass tube at 0 ºC for 0.5 h (Figure 3). Then the ice-bath and ammonia-gas line inlet are removed and the reaction mixture is stirred at room temperature for 2 h under air with the precipitated
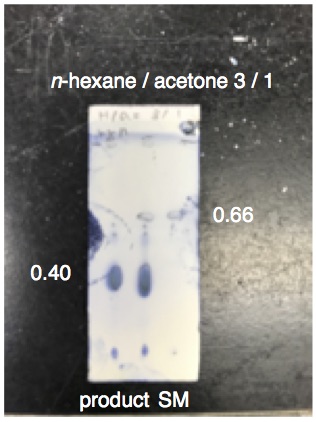
ammonium chloride (Figure 4). The reaction progress is monitored by TLC analysis (Figure 5) (
Note 7). The solvent is removed using a rotary evaporator (30 °C, 64 mmHg). The resulting crude product containing
2 and
ammonium chloride is placed on a sintered glass filter, which is washed with
chloroform (3 x 20 mL) (
Note 8) under reduced pressure into a 300-mL one-necked, round-bottomed flask (Figure 6). The filtrate is concentrated using a rotary evaporator (30 °C, 64 mmHg), and the flask is fitted with a 2.5-cm Teflon-coated oval magnetic stirring bar. The crude solid product is dissolved in
chloroform (40 mL), and heated in an oil bath at 70 ºC (bath temperature).
n-Hexane (70 mL) is slowly added to the hot
chloroform solution (
Note 9). The solution is allowed to cool to room temperature and left standing for 4 h, and then cooled in a refrigerator (4 ºC) for 24 h. The resulting crystals are collected by filtration with a Hirsch funnel (Figure 7) and washed with a mixed solvent of
n-hexane and
CHCl3 (5/1, 3 x 3 mL). The collected crystals are dried in vacuo (60 ºC, 0.1 mmHg) for 5 h to provide 7.84 g of phosphinothioic amide
2 (77% yield) as colorless crystals (Figure 8) (Notes
10 and
11). The purity of phosphinothioic amide
2 is assessed at >98 wt% by quantitative
1H NMR in CDCl
3 using
dimethyl fumarate as a standard (
Note 12).

|
|
|
Figure 1. Reaction setup Step A
|
Figure 2. TLC image.
|
|

|

|
|
Figure 3. Addition of ammonia
|
Figure 4. Precipitated ammonium chloride
|
|
|

|
|
Figure 5. TLC image 2
|
Figure 6. After filtration
|
|

|

|
|
Figure 7. Filtration
|
Figure 8. Phosphinothioic amide 2
|
|
B.
P,P-Diphenyl-N-(1-phenylethylidene)phosphinothioic amide (
4).
4 A 50 mL one-necked round-bottomed flask equipped with a 2.5-cm Teflon-coated oval magnetic stirring bar is charged with phosphinothioic amide
2 (7.00 g, 30 mmol, 1.0 equiv) and
(1,1-dimethoxyethyl)benzene (
3) (4.99 g, 30 mmol, 1.0 equiv) (
Note 13). The flask is equipped with a reflux condenser (15 cm) without tap water flow (air-cooling), and the mixture is heated to 130 °C (oil bath temperature) under air (Figures 9 and 10). After stirring for 1 h, another portion of acetal
3 (4.99 g, 30 mmol, 1.0 equiv) (
Note 13) is added dropwise at that temperature. After further stirring for 1 h, the mixture is cooled to room temperature (Figure 11), the reaction progress is checked by TLC analysis (
Note 14). The mixture is transferred to a 300 mL one-necked round-bottomed flask using 15 mL
EtOAc (
Note 15) and 20 g neutral silica gel (
Note 16) is added. The mixture is concentrated first by a rotary evaporator (30 °C, 64 mmHg) and then under high vacuum (rt, 0.2 mmHg) for 40 min (Figure 12). The crude mixture is applied on top of the silica gel column (a glass column charged with 7 x 10 cm, 210 g of neutral silica gel) (
Note 16), and eluted with 550 mL of a mixed solvent of
EtOAc and
n-hexane (1/10) (Notes
9 and
15), followed by 500 mL of a mixed solvent of
EtOAc and
n-hexane (1/5). At this point, collection of the eluent is begun (100-mL fractions each), and collection is continued while using 1.9 L of a mixed solvent of
EtOAc/
n-hexane (1/5). The product is contained in fractions 2-17, which are concentrated using a rotary evaporator (65 mmHg, 30 °C). The residue is dissolved in 50 mL hot
EtOAc (50 °C), and 150 mL
n-hexane is added (Notes
9 and
15). The solution is allowed to cool to room temperature, and further cooled in a refrigerator (4 °C) for 24 h. The resulting crystals are collected by suction filtration on a Hirsch funnel (Figure 13), washed with 100 mL
n-hexane, and transferred to a 50-mL vial and dried in vacuo (rt, 0.2 mmHg) for 4 h to provide 2.64 g of ketimine
4 (26% yield) as colorless crystals (Figure 14) (Notes
17 and
18). The purity of ketimine
4 is assessed at >98 wt% by quantitative
1H NMR in CDCl
3 using
dimethyl fumarate as a standard (
Note 12).

|

|
|
Figure 9. Reaction setup Step B
(Before heating)
|
Figure 10. Reaction setup Step B
(while heating at 130 ºC)
|
|

|

|
|
Figure 11. After cooling
|
Figure 12. After vacuum
|
|

|

|
|
Figure 13. Filtration
|
Figure 14. Purified product 4
|
|
C.
(R)-Diethyl (1-((diphenylphosphorothioyl)amino)-1-phenylethyl)phosphon-ate (
6).
5 An oven-dried 20 mL two-necked round-bottomed flask equipped with a 1.5-cm Teflon-coated oval magnetic stirring bar, 3-way glass stopcock with an argon balloon, and a rubber septum is charged with
1,2-bis((2R,5R)-2,5-diphenylphospholano)ethane (
(R,R)-Ph-BPE, 122 mg, 0.24 mmol, 0.02 equiv) (
Note 19) and
tetrakis(acetonitrile)copper(I) hexafluoro-phosphate (
[Cu(CH3CN)4]PF6, 89.5 mg, 0.24 mmol, 0.02 equiv) (
Note 20). The flask is evacuated and backfilled with an argon (3 times). Dry
THF (2 mL) (
Note 4) is added under an argon atmosphere (
Note 21), and the mixture is stirred at room temperature for 1.5 h to give a solution of the chiral Cu(I) complex, which is stored at room temperature and used within 1 h.
An oven-dried 100-mL two-necked round-bottomed flask equipped with a 2.5-cm Teflon-coated oval magnetic stirring bar, a 3-way glass stopcock with an argon balloon, and a rubber septum is charged with
P,P-diphenyl-N-(1-phenylethylidene)phosphinothioic amide (
4) (4.03 g, 12 mmol, 1.0 equiv). The flask is evacuated (rt, 5 min) and backfilled with argon gas (3 times) (Figure 15). Dry
THF (44 mL) (
Note 4) is added via a syringe under an argon atmosphere. The mixture is stirred for 2 min to give a clear solution (Figure 16). The catalyst solution (2.0 mL) containing Cu (I) complex (0.24 mmol, 0.02 equiv), which is prepared in the procedure described above, is transferred (using 4.0 mL dry
THF for rinsing) via a syringe at room temperature under argon atmosphere (Figure 17). After stirring for 5 min,
triethylamine (836 µL, 6 mmol, 0.5 equiv) (
Note 22) is added via a syringe under an argon atmosphere.
Diethyl phosphite (
5) (2.94 mL, 24 mmol, 2.0 equiv) (
Note 23) is added via a syringe under an argon atmosphere (Figure 18). The flask is evacuated and backfilled with argon gas (3 times), and attached to nitrogen-gas line inlet (Figure 19). After stirring at room temperature for 72 h under continuous nitrogen flow (Figure 20), the reaction progress is checked by TLC analysis (
Note 24). To the mixture is added neutral silica gel (10 g) (
Note 16) and the volatiles are removed under reduced pressure (80 mmHg, 40 °C). The resulting residue is applied to the top of silica gel column (a glass column charged with 7 x 10 cm, 210 g of neutral silica gel), and eluted with 600 mL of a mixed solvent of
EtOAc/
n-hexane (1/2) (Notes
9 and
15), followed by 1.0 L of a mixed solvent of
EtOAc/
n-hexane (2/3). At this point, collection of eluents (100 mL fractions) is begun and continued with 2.5 L of a mixed solvent of
EtOAc/
n-hexane (2/3). The desired product is contained in fractions 6-26, which are concentrated using a rotary evaporator (80 mmHg, 40 °C). The resulting residue is transferred to a 50-mL round-bottomed flask using
EtOAc (30 mL) (
Note 15), and dried in vacuo (0.1 mmHg at 90 °C for 12 h) to provide 5.02 g of phosphonate
6 (88% yield) as a brown viscous oil (Figures 21 and 22). The purity of phosphonate
6 is assessed at >98 wt% by quantitative 1H NMR in CDCl3 using
dimethyl fumarate as a standard (
Note 12), and the enantiopurity of 6 is determined to be 95% ee by HPLC analysis (Notes
25,
26, and
27).

|

|
|
Figure 15. Reaction setup Step C(charged starting material)
|
Figure 16. Reaction setup Step C(dissolved starting material)
|
|

|

|
|
Figure 17. Reaction setup Step C(after addition of catalyst)
|
Figure 18. Reaction setup Step C(after addition of diethyl phosphite)
|
|

|

|
|
Figure 19. Reaction setup Step C(under nitrogen flow)
|
Figure 20. Reaction setup Step C(after 72 h of stirring)
|
|

|

|
|
Figure 21. Purified product 6(before drying)
|
Figure 22. Purified product 6(after drying)
|
|
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical). See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
chlorodiphenylphosphine,
sulfur,
tetrahydrofuran,
ammonia,
ammonium chloride,
chloroform,
n-hexane,
dimethyl fumarate,
(1,1-dimethoxyethyl)benzene,
ethyl acetate, silica gel,
1,2-bis((2R,5R)-2,5-diphenylphospholano)ethane,
tetrakis(acetonitrile)copper(I) hexafluoro-phosphate,
triethylamine, and
diethyl phosphite.
2.
Chlorodiphenylphosphine (96%) was purchased from Sigma-Aldrich Co., Inc. and used as received.
3.
Sulfur (99%, reagent grade) was purchased from Sigma-Aldrich Co., Inc. and used as received.
4. Dry
THF (>99.5%) was purchased from Kanto Chemical Co., Inc., which was purified under argon by using an Organic Solvent Pure Unit (Wako Pure Chemical Industries, Ltd.).
5. TLC plates were purchased from Merck Millipore, Co., Inc. (silica gel 60 F254).
6. During the sulfurization, several spots appeared on TLC, caused by decomposition of
P,P-diphenyl-phosphinothioic chloride, (R
f 0.91 and 0.80,
chloroform/
acetone = 9/1). The checkers judged the endpoint of sulfuration with
31P NMR (162 MHz, CDCl
3), confirming disappearance of the peak of
chlorodiphenylphosphine (
1) (δ 81.8 ppm) and appearance of the peak of
P,P-diphenyl-phosphinothioic chloride (δ 80.0 ppm). The sampling was performed by taking a 10-µL fraction of the reaction mixture using a gas-tight syringe, which was diluted with CDCl
3 (0.7 mL) in an NMR tube.
7. The amination progress was monitored by TLC analysis (
n-hexane/
acetone = 3/1) using ceric ammonium molybdate stain (Figure 5). The phosphinothioic amide
2 appeared at R
f = 0.40 (blue).
8.
Chloroform (>99.0%) was purchased from Kanto Chemical Co., Inc. and used as received.
9.
n-Hexane (96%) was purchased from Wako Pure Chemical Industries and used as received.
10. A second reaction on the same scale provided 8.10 g (77%) of
2, and a third reaction on the same scale gave 7.84 g (74%) of
2.
11.
Phosphinothioic amide, P,P-diphenyl (
2) is bench-stable and has the following spectroscopic properties: R
f 0.40 (
n-hexane/
acetone = 3/1); mp 108-109 ºC;
1H NMR
pdf(400 MHz, CDCl
3) δ: 2.90 (brs, 2H), 7.42-7.49 (m, 6H), 7.96-8.00 (m, 4H);
13C NMR
pdf(150 MHz, CDCl
3) δ: 128.4, 128.5, 131.1, 131.2, 131.6, 131.7, 134.8, 135.5;
31P NMR (162 MHz, CDCl
3) δ: 55.4; IR (
CHCl3):
ν 3344, 3228, 3106, 3054, 1551, 1475, 1440, 1306 cm
-1; HRMS (ESI-TOF) Anal. calcd. for C
12H
13NPS
m/z 234.0501 [M+H]
+, found 234.0503.
12.
Dimethyl fumarate (>98%) was purchased from Tokyo Chemical Industry (TCI, product number F0069) and used as received.
13.
(1,1-Dimethoxyethyl)benzene (97%) was purchased from Alfa Aesar Co., Inc. and used as received.
14. The reaction progress was monitored by TLC analysis (
EtOAc/
n-hexane, 1/5) using ceric ammonium molybdate or potassium permanganate stain (see Figure). The starting material
2 appeared at R
f = 0.15 (blue with ceric ammonium molybdate), and the ketimine
4 appeared at R
f = 0.45 (blue with ceric ammonium molybdate).
15.
EtOAc (99%) was purchased from Wako Pure Chemical Industries and used as received.
16. Neutral silica gel was purchased from Kanto Chemical Co., Inc. (silica gel 60N; spherical, 40-50 µm).
17. A second reaction on the same scale provided 3.40 g (34%) of
4, and a third reaction on the same scale gave 4.13 g (41%) of
4.
18.
P,P-Diphenyl-N-(1-phenylethylidene)phosphinothioic amide (
4) is bench-stable and has the following spectroscopic properties: R
f 0.45 (
EtOAc/
n-hexane = 1/5); mp 129-131 ºC;
1H NMR
pdf(600 MHz, CDCl
3) δ: 2.80 (d, 3H,
J = 1.5 Hz), 7.42-7.49 (m, 8H), 7.54-7.57 (m, 1H), 8.03-8.07 (m, 6H);
13C NMR
pdf(150 MHz, CDCl
3) δ: 22.4 (d,
J = 17 Hz), 127.9, 128.3, 128.4, 128.5, 131.08, 131.11, 131.2, 132.5, 136.1, 136.8, 139.9, 140.1, 183.4 (d,
J = 8.3 Hz);
31P NMR (162 MHz, CDCl
3) δ: 47.4; IR (
CHCl3):
ν 3048, 1626, 1591, 1574, 1475, 1434, 1370, 1307, 1266 cm
-1; HRMS (ESI-TOF) Anal. calcd. for C
20H
19NPS
m/z 336.0970 [M+H]
+, found 336.0973.
19.
(-)-1,2-Bis((2R,5R)-2,5-diphenylphospholan-1-yl)ethane (
(R,R)-Ph-BPE, Kanata purity) was purchased from Sigma-Aldrich Co., Inc. and used as received.
20.
Tetrakis(acetonitrile)copper(I) hexafluorophosphate (
[Cu(CH3CN)4]PF6) (97%) was purchased from Sigma-Aldrich Co., Inc. and used as received.
21. Although the use of a dry box is recommended,
(R,R)-Ph-BPE and
[Cu(CH3CN)4]PF6 can be quickly weighed under air.
22.
Triethylamine (99%) was purchased from Wako Pure Chemical Industries and distilled from calcium hydride before use.
23.
Diethyl phosphite (>98%) was purchased from Tokyo Chemical Industry and distilled under vacuum (4.0 mmHg, 50 °C) before use.
24. The reaction progress was monitored by TLC analysis (
EtOAc/
n-hexane = 2/1) using ceric ammonium molybdate or potassium permanganate stain (see Figure). The ketimine starting material
4 appeared at R
f = 0.90 (blue), and the hydrophosphonylation product appeared at R
f = 0.40 (blue) using ceric ammonium molybdate stain. The
diethyl phosphite starting material
5 appeared at R
f = 0.30 (yellow) using potassium permanganate stain.
25. A second reaction on the same scale provided 4.41 g (78%) of
6, and a third reaction on the same scale gave 5.28 g (93%) of
6.
26.
(R)-Diethyl-(1-((diphenylphosphorothioyl)amino)-1-phenylethyl) phosphonate (
6) is bench-stable and has the following spectroscopic properties: R
f 0.40 (
EtOAc/
n-hexane = 2/1);
1H NMR
pdf(600 MHz, CDCl
3) δ: 1.09 (t, 1H,
J = 7.0 Hz), 1.31 (t, 1H,
J = 7.0 Hz), 1.98 (d, 3H,
J = 17.3 Hz), 3.51-3.55 (m, 1H), 3.80-3.84 (m, 1H), 3.95 (t, 1H,
J = 7.6 Hz), 4.06-4.14 (m, 2H), 7.28-7.30 (m, 1H), 7.34-7.39 (m, 4H), 7.43-7.52 (m, 4H), 7.56-7.57 (m, 2H), 7.80-7.84 (m, 2H), 8.16-8.20 (m, 2H);
13C NMR
pdf(150 MHz, CDCl
3) δ: 16.2 (d,
J = 5.5 Hz), 16.4 (d,
J = 6.1 Hz), 19.8 (t,
J = 5.1 Hz), 58.7, 59.7, 63.2 (d,
J = 7.7 Hz), 63.7 (d,
J = 7.1 Hz), 127.37, 127.40, 127.49, 127.51, 127.83, 127.85, 128.1, 128.2, 128.4, 128.5, 130.86, 130.94, 131.45, 131.47, 131.49, 131.50, 132.1, 132.2, 135.5, 135.7, 136.2, 136.4, 140.3;
31P NMR (162 MHz, CDCl
3) δ: 53.1 (d,
J = 40 Hz), 24.5 (d,
J = 40 Hz); IR (
CHCl3):
ν 3373, 3048, 2979, 2926, 2903, 1496, 1481, 1440, 1394, 1237 cm
-1; HRMS (ESI-TOF) Anal. calcd. for C
24H
29O
3NaP
2S
m/z 496.1236 [M+Na]
+, found 496.1242; [α]
D24 46.5 (
c 1.00,
CHCl3); Enantiomeric excess of the product was determined to be 95% ee by HPLC analysis on chiral stationary phase (CHIRALPAK IA (
Φ 0.46 cm x 25 cm), 2-propanol/
n-hexane = 1/9, flow rate 1.0 mL/min, detection at 254 nm, t
R = 9.6 min (minor), 12.8 min (major)).
27. The Cu(I) catalyst (2 mol%, 0.02 equiv) and
triethylamine (50 mol%, 0.5 equiv) were used for better reproducibility in terms of enantioselectivity on a >5 g scale reaction.
3. Discussion
In this context, a pioneering catalytic asymmetric process for the synthesis of nonracemic α,α-disubstituted α-amino phosphonic acids was reported by Ito et al. by enantioselective allylation of α-acetoamido-β-keto phosphonates, although the enantioselectivity was not satisfactory.
7 Nakamura and Shibata et al. later reported highly enantioselective hydrophosphonylation of secondary phosphites to
N-sulfonyl ketimines promoted by cinchona alkaloids to afford this class of compounds.
8,9 Although this catalytic system exhibited broad generality for aromatic
N-sulfonyl ketimines and aliphatic substrates gave lower enantioselectivity, Shibasaki et al. later devised a cooperative catalytic system to render a highly enantioselective reaction of both aromatic and aliphatic ketimines.
5 The use of soft Lewis basic
N-thiophosphinoylketimines was key to promote the reaction with the soft Lewis acid (chiral Cu(I) complex)/Brønsted base (Et
3N) cooperative catalyst. In small scale reactions, as little as 0.5% of catalyst loading was sufficient to reach completion and the chiral Cu(I) complex could be recovered. For large scale reactions, the use of 2 mol% of catalyst is recommended to ensure high enantioselectivity.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Phosphinothioic amide, P,P-diphenyl- (2) (17366-80-2)
Chlorodiphenylphosphine (1) (1079-66-9)
Sulfur (7704-34-9)
Tetrahydrofuran (109-99-9)
Ammonia (7664-41-7)
Ammonium chloride (12125-02-9)
Dimethyl fumarate (624-49-7)
Phosphinothioic chloride, P,P-diphenyl- (1015-37-8)
(1,1-Dimethoxyethyl)benzene (3) (4316-35-2)
P ,P-Diphenyl-N-(1-phenylethylidene)phosphinothioic amide (4) (945492-04-6)
1,2-Bis((2R,5R)-2,5-diphenylphospholano)ethane (528565-79-9)
Tetrakis(acetonitrile)copper(I) hexafluorophosphate (64443-05-6)
Triethylamine (121-44-8)
Diethyl phosphite (5) (762-04-9)
(R)-Diethyl (1-((diphenylphosphorothioyl)amino)-1-phenylethyl)phosphonate (6) (1446718-96-2)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved