Org. Synth. 2021, 98, 147-170
DOI: 10.15227/orgsyn.098.0147
Synthesis of the Isocyanide Building Block Asmic, anisylsulfanylmethylisocyanide
Embarek Alwedi, Bilal Altundas, Allen Chao, Zachary L. Ziminsky, Maanasa Natrayan, and Fraser F. Fleming*
1Checked by Names Anthony J. Fernandes, Martina Drescher, and Nuno Maulide
1. Procedure (Note 1)
A. N-((2-Methoxyphenylthio)methyl)formamide (1). A 3-necked, 500 mL round-bottomed flask (Note 2), equipped with a magnetic stir bar (PTFE-coated, cylindrical, 3 cm) (Figure 1a) is charged with paraformaldehyde (17.7 g, 584 mmol, 4.1 equiv), formamide (43 mL, 48.6 g, 1.08 mol, 7.6 equiv), formic acid (27 mL, 33 g, 713 mmol, 5 equiv), and 2-methoxybenzenethiol (17.4 mL, 20 g, 142.6 mmol, 1 equiv) (Note 3) (Figure 1b). The left and right necks are stoppered with a rubber septum and the middle neck is connected to a reflux condenser stoppered with a rubber septum that is pierced with a 20-gauge disposable needle open to the atmosphere (Figure 2).
Figure 1. (A) Flask with stir bar; (B) Flask with stir bar and reagents (photos provided by authors)
Figure 2. Reaction set-up prior to heating
(photo provided by submitter)
The flask is immersed in an oil bath that is gradually heated to 100 °C over 1 h; the temperature is then maintained at 100 °C for an additional 3 h (Note 4) (Figure 3a-c).
Figure 3. (A) Reaction mixture at 91 °C; (B) Reaction mixture at 100 °C; (C) Reaction mixture after 3h at 100 °C
(photos provided by submitters)
The reaction is monitored by silica gel thin layer chromatography with both 5% (5:95) EtOAc-hexanes as the eluent to check for the presence of 2-methoxybenzenethiol and 75% (3:1) EtOAc-hexanes as eluent to monitor the formation of the formamide 1. The TLC plate is visualized with UV light (Note 5) (Figure 4a). The reaction mixture is then allowed to cool to room temperature (Figure 4b).
Figure 4. (A) TLC analysis after 3 h at 100 °C; (B) Reaction mixture after cooling to rt
(photos provided by submitters)
Figure 5. (A) The reaction mixture after the addition of water; (B) The reaction mixture after the addition of dichloromethane
(photos provided by submitters)
Distilled water (200 mL) and dichloromethane (80 mL) are added to the crude reaction mixture (Note 6) (Figure 5). The contents of the flask are transferred to a 1 L separatory funnel (Figure 6a). The phases are separated, and then the aqueous phase is extracted with dichloromethane (3 x 100 mL). The combined organic extract is washed with brine (200 mL) (Figure 6b), dried for 10 min over anhydrous sodium sulfate (Na2SO4, 80 g) and concentrated to afford the crude formamide as a clear, pale yellow viscous oil (Figure 6c). The formamide is purified by precipitation from a dichloromethane solution through the addition of hexane. The crude formamide is dissolved in dichloromethane (95 mL) to which hexanes is added portion-wise until no further precipitation is observed (Note 7) (Figure 7). The resultant suspension is placed in a -20 °C freezer.

Figure 6. (A) Crude reaction mixture; (B) The crude reaction mixture after aqueous washing with brine; (C) Crude formamide (photos provided by submitters)
Figure 7. (A) Dissolution of the crude formamide in CH2Cl2; (B) Addition of 100 mL of hexanes; (C) Addition of a further 50 mL of hexanes -150 mL total; (D) Addition of a further 50 mL of hexanes -200 mL total; (E) Addition of a further 50 mL of hexanes -250 mL total (photos provided by submitters)
After 19 h, the white solid (Figure 8a) is isolated by filtration through a 500 mL medium porosity, sintered Büchner funnel (Figure 8b). The solid on the frit is washed with cold (0 °C) hexanes (2 x 25 mL). The solid is collected, dissolved in CH2Cl2 (50 mL) and then precipitated with hexanes (200 mL) (Note 8). The white solid is filtered and then washed with cold (0 °C) hexanes (2 x 25 mL). The solid is again collected, dissolved in CH2Cl2 (50 mL), precipitated with hexanes (200 mL), filtered, and washed with cold (0 °C) hexanes (2 x 25 mL). The white solid is then dried under vacuum (1 mmHg) for 1 h at room temperature to afford 19.0 g (68% yield) of formamide 1 as a white solid (Figure 8c) (Notes 10, 11, and 12).
Figure 8. (A) Formamide 1 after 19 h at -20 °C (B) Formamide 1 after filtration (C) Formamide after third precipitation (submitters); (D) Formamide after third precipitation (checkers)
B. Anisylsulfanylmethylisocyanide (Asmic (2). A 3-necked, 500 mL round-bottomed flask, equipped with a magnetic stir bar (PTFE-coated, cylindrical, 3 cm) (Note 2) is charged with N-((2-methoxyphenylthio)methyl)formamide (18.5 g, 93.8 mmol, 1 equiv) (Note 9) (Note 13). The left and right necks of the flask are stoppered with rubber septa (Figure 9a). Dichloromethane (108 mL) (Note 14) is added at room temperature (20 °C) (Figure 9b). After the formamide is completely dissolved, diisopropylethylamine (81.7 mL, 60.6 g, 469.0 mmol, 5 equiv) (Note 15) is added from a graduated cylinder through the middle neck (Note 16).
Figure 9. (A) Reaction set-up with formamide 1; (B) Dissolution of formamide 1 in CH2Cl2; (C) Reaction mixture after addition of diisopropylethylamine
(photos provided by submitters)
After 10 min, the middle neck is equipped with a 100 mL, pressure-equalizing addition funnel capped with a septum pierced with a 20-gauge needle open to the atmosphere (Figure 10). The two side necks are capped with septa that are each pierced with a 20-gauge needle open to the atmosphere. The reaction mixture is immersed in an ice-salt bath that is cooled to -10 °C (Note 17) (Figure 10a). The addition funnel is charged with POCl3 (16.4 mL, 25.8 g, 188.0 mmol, 2 equiv) (Note 18) that is added dropwise, over 20 min; the reaction temperature is maintained between -12 and -9 °C during the addition (Note 19) (Figures 10b and 10c).
Figure 10. (A) Reaction mixture at -12 °C with POCl3 charged in the addition funnel; (B) Reaction mixture 20 min after the addition of POCl3; (C) Reaction mixture 90 min after the addition of POCl3
(photos provided by submitters)
The reaction is monitored by thin layer chromatography (TLC) on silica gel with both 10% (1:9) EtOAc-hexanes and 75% (3:1) EtOAc-hexanes as eluent and visualized under UV light (Note 20) (Figure 11). After 1.5 h, TLC analysis indicated that the reaction is complete.
Figure 11. (A) TLC analysis of the reaction after 1.5 h; (B) The crude reaction mixture after 1.5 h
(photos provided by submitters)
The light brown reaction mixture is diluted with dichloromethane (95 mL), transferred to a 2-L Erlenmeyer flask equipped with a magnetic stir bar (PTFE-coated, cylindrical, 3 cm) and then cooled in an ice bath (Figure 12a). A cold (0 °C), saturated, aqueous solution (500 mL) of NaHCO3 is slowly added down the side of the flask via pipette into the stirred reaction mixture until the initially vigorous reaction subsided; the remaining NaHCO3 solution is added by slowly pouring the remaining NaHCO3 solution into the reaction flask (Note 21) (Figure 12b) until the solution reached a pH = 8 (Note 22) (Figure 12c).
Figure 12. (A) Reaction mixture after dilution with CH2Cl2; (B) Reaction mixture after neutralization; (C) The pH after addition of saturated, aqueous NaHCO3
(photos provided by submitters)
The crude reaction mixture is allowed to warm to room temperature (Figure 13a) and then transferred to a 1-L separatory funnel (Figure 13b). The phases are separated (Note 23), the organic layer is collected, and the aqueous phase further extracted with dichloromethane (95 mL) (Figure 13c).
Figure 13. (A) Crude reaction mixture at room temperature; (B) Reaction mixture after transfer to the separatory funnel; (C) CH2Cl2 extract of the aqueous phase
(photos provided by submitters)
The combined organic phase is concentrated by rotary evaporation (30 ° C, 20 mm Hg) to give an amber oil (Figure 14a). The crude oil is dissolved in ethyl acetate (370 mL) and then washed with brine (4 x 230 mL) (Figures 14b and c).
Figure 14. (A) Appearance of crude reaction mixture after concentration; (B) Dissolution of crude Asmic in EtOAc followed by washing with brine; (C) Crude Asmic after the fourth wash with 250 mL brine
(photos provided by submitters)
The phases are separated and then the organic phase is combined (Figure 15a). The crude solution of Asmic (2) is dried for 10 min over anhydrous Na2SO4 (37 g, Figure 15b), filtered (Figure 15c) and then concentrated by rotary evaporation (30 °C, 20 mmHg). Crude Asmic is subjected to a high vacuum (20 °C, 0.1 mmHg) for 15 min, which gives a viscous amber oil in 17.1 g, 101% yield (Figure 15d, Note 24). Asmic (2) obtained at this stage is sufficiently pure for many applications without requiring further purification.
Figure 15. (A) The combined organic phase after extraction; (B) The dried organic phase over Na2SO4; (C) The dry, filtered organic phase; (D) Crude Asmic after removal of the solvent (photos provided by submitters)
Asmic (2) (17.1 g) is dissolved in CH2Cl2 (95 mL) and 21 g Celite (21 g) is added (Note 25). The solvent is removed by rotary evaporation (30 ° C, 20 mmHg), followed by high vacuum (20 °C, 0.1 mmHg) for 30 min. The adsorbed material (Figure 16) is charged on a column (6 x 12 cm) of 118 g of silica gel (Note 26) pre-equilibrated with heptane (Figure 17), and the column is eluted with 2.2 L of 10% ethyl acetate in n-heptane (1:9 Ethyl acetate:n-heptane). After 450 mL of solvent elutes, 25 mL fractions are collected, and the elution continues for 56 fractions. Pure Asmic (2) is obtained in fractions 16-77 (Figure 18), which are concentrated by rotary evaporation (30 °C, 20 mmHg) and then under high vacuum (20 °C, 0.1 mmHg) for 15 min to afford a clear oil. The oil is stored in a -20 °C freezer for 12 h resulting in 14.2 g (85% yield) of an off-white crystalline solid (Notes 27, 28, 29, and 30) (Figure 19). Asmic (2) is stable for at least 9 months when stored at -20 °C.

Figure 16. Asmic (2) adsorbed on Celite (photo provided by submitters)
Figure 17. Silica gel column setup with adsorbed Asmic (2) (photo provided by submitters)
Figure 18. TLC of fractions 7-53 containing product (2) (photo provided by submitters)
Figure 19. (A) Off-white crystalline Asmic (2) after rotary evaporation, high vacuum, and storage in a -20 °C freezer for 12 h (submitters); (B) crystalline Asmic (2) (checkers)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
paraformaldehyde,
formic acid,
formamide,
2-methoxybenzenethiol,
N,N-diisopropylethylamine,
phosphorous oxychloride,
sodium hydrogen carbonate,
hexane,
dichloromethane,
ethyl acetate,
heptane, silica gel, and Celite, as well as the proper procedures for solvent evaporation and the application of high vacuum.
2. All glassware was dried in an oven (120 °C) for 1 h prior to use.
3.
Paraformaldehyde (96%),
formic acid (97%),
formamide (high purity) and
2-methoxybenzenethiol (95%) were purchased from Acros, Alfa Aesar, VWR, and Oakwood Chemicals, respectively, and used as is without purification.
Formamide and
formic acid are used in excess as they serve as the solvent for the reaction. The Process Mass Intensity, PMI, is 71, considerably less than the industry standard of 200. The checkers purchased
formamide (≥99%) and
2-methoxybenzenethiol (97%) from Sigma Aldrich.
4. Upon heating, the reaction mixture changed from an initial white heterogeneous suspension of
paraformaldehyde (Figure 3a) to a yellow suspension (Figure 3b) and then to a clear yellow solution after 3 h at 100 °C (Figure 3c). Upon cooling to rt, a clear, very pale yellow solution of
formamide 1 is formed (Figure 4d).
5. An aliquot (0.1 mL) is removed and diluted with
CH2Cl2 (3 mL) to analyze the reaction mixture via TLC.
6. The reaction mixture may appear white and cloudy due to some precipitation of the desired
formamide. If significant precipitation is observed, the solid can be filtered and the remaining solution subjected to the precipitation procedure described. The best result is obtained when the reaction mixture is diluted with
dichloromethane and washed as described.
7. Portion-wise addition of
hexane is recommended as rapid addition of
hexane sometimes results in the formation of an oil that is unable to be precipitated.
8. The first recrystallization results in a white powder that may require iterative recrystallizations to obtain white crystalline solid
formamide.
9. The material was dried for 1 h under high vacuum (0.1 mmHg) before performing the dehydration.
10. A second run by the checkers was performed at half scale and provided 12.3 g (87%) of the product.
11.
N-((2-Methoxyphenylthio)methyl)formamide was obtained as an off-white, crystalline solid. The sample presented the following analytical data: mp 82-84 ℃; IR (neat) cm
-1 3285, 3059, 2939, 2837, 1664, 1477, 1243, 751. At 25 °C, two rotamers, one major and one minor, of the
formamide are observed in the NMR spectra:
1H NMR
pdf (700 MHz, CDCl
3) δ: 3.89 (s, 1H), 3.91 (s, 4H), 4.55 (d,
J = 7.0 Hz, 1H), 4.69 (d,
J = 6.1 Hz, 2H), 5.94 (s, 1H), 6.04 (s, 1H), 6.95 - 6.89 (m, 3H), 7.32 - 7.27 (m, 1H), 7.35 (dd,
J = 11.0, 4.5 Hz, 1H), 7.44 - 7.38 (m, 2H), 7.74 (d,
J = 11.7 Hz, 1H), 8.10 (s, 1H).
13C NMR
pdf (176 MHz, CDCl
3) δ : 41.0, 46.3, 56.0, 56.1, 111.3, 111.5, 119.1, 121.1, 121.4, 121.5, 129.7, 131.0, 133.5, 136.4, 158.7, 159.6, 160.6, 163.8. HRMS (
m/z) calcd for C
9H
11NNaO
2S [M+Na]
+ : 220.0403, found: 220.0399.
12. The precipitated
formamide was used directly for the synthesis of
Asmic. The submitters reported the following crystallization procedure:
Formamide (24 g) is dissolved in 50 mL of hot (60 °C) isopropyl alcohol, which is then allowed to cool to room temperature (22 °C) over 12 h to afford 14.1 g (50% yield) of an off-white, crystalline solid. The checkers assessed purity before and after recrystallization by qNMR
pdf using 1,3,5- trimethoxybenzene as an internal standard. Purity was assessed as 78% prior to crystallization and 83% post-crystallization.
13. The submitters started with 20 g, (101 mmol) of
N-((2-methoxyphenylthio)methyl)formamide, owing to their larger yield (75%) for the previous step.
14.
Dichloromethane (ACS grade) was purchased from Fisher Scientific and used as is without purification.
15.
N,N-Diisopropylethylamine (99%) was purchased from Oakwood Chemicals and used as is. Excess
N,N-diisopropylethylamine was employed to minimize hydrolysis by maintaining a basic medium throughout.
16. Precipitation may be observed after addition of
N,N-diisopropylethylamine, but this is not detrimental to the overall reaction.
17. The temperature is maintained below -9 °C during the course of the reaction.
18.
Phosphorus oxychloride (
POCl3, 99%) was purchased from ACROS Organics and used as is without purification.
19. The addition commenced slowly at -12 °C to modulate a rapid exotherm that results in an initial, slight elevation in the reaction temperature. The temperature was maintained between -12 °C and -9 °C throughout the addition.
20. An aliquot (0.2 mL) was removed from the reaction mixture, diluted with
CH2Cl2 (3 mL) and carefully quenched with saturated aqueous
NaHCO3 solution. The mini-workup provided a clear visualization of the reaction mixture which avoids streaking due to the excess amine.
21. The saturated aqueous
NaHCO3 was added dropwise to control a rapid exotherm that occurred upon the initial addition. Once the initial exotherm subsides, the saturated, aqueous
NaHCO3 solution can be slowly added by pouring the solution down the side of the flask. The pH was determined by testing with pH paper.
22. The pH of the mixture should not be higher than pH = 9 as this can sometimes result in the detrimental basic hydrolysis of the
isocyanide to the
formamide.
23. Care should be taken during the extraction; the separatory funnel should be gently shaken to avoid emulsions and the risk of a pressure build up from the evolution of CO
2.
24. The submitters reported 18.05 g (99%, based on their larger scale) of the product prior to column chromatography.
25. Celite
TM 545 (CAS # 68855-54-9) was purchased from Millipore Sigma and used as is.
26. Silica gel (SiliaFlash P60, 40-63 μm (230-400 mesh), 60 Å) was purchased from Silicycle and used as is.
27. A second run on half-scale provided 7.65 g (84%) of
Asmic.
28. Analytical data for
Asmic (2): mp 27-29 °C; IR (neat) cm
-1 3065, 2969, 2940, 2837, 2137, 1581, 1477, 1243, 1021, 749.
1H NMR
pdf (600 MHz, CDCl
3) δ: 3.92 (s, 3H), 4.58 (s, 2H), 6.94 (d,
J = 8.3 Hz, 1H), 6.99 (td,
J = 7.5, 1.0 Hz, 1H), 7.38 (td,
J = 8.2, 1.7 Hz, 1H), 7.53 (dd,
J = 7.6, 1.6 Hz, 1H).
13C NMR
pdf (151 MHz, CDCl
3) δ: 42.9, 42.9, 43.0, 56.0, 111.3, 118.8, 121.5, 131.0, 134.9, 158.9 (2C, broad). HRMS (
m/z) calcd for C
9H
9NNaOS [M+Na]
+ : 202.0297. Found: 202.0295.
29. Purity of
Asmic (
2) (17.9 mg) was assessed as 99.5 wt% using qNMR
pdf with 1,3,5-trimethoxybenzene (16.8 mg) as an internal standard. No recrystallization was performed.
30. The submitters report that
Asmic (2) can be recrystallized from CCl
4 and pentane:
Asmic (5.76 g) was dissolved in CCl
4 (8 mL) at rt and then was diluted with small volume of pentane and then cooled, initially to 0 °C and then to -20 °C. Additional pentane was added to initiate crystallization and the solution left at -20 °C for 3 days. The crystals were filtered, washed three times 5 mL portions of CCl
4/pentane (1:5), and then dried under high vacuum at rt to afford 4.04 g (73%) of pure
Asmic as a colorless crystalline solid. Concentration and recrystallization of the filtrate afforded additional pure
Asmic (
2).
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Isocyanides are extremely important precursors for a diverse suit of bond forming reactions: multi-component reactions,
2 transition metal insertions,
3 and radical reactions.
4 The promiscuous reactivity, driven by conversion of the terminal, divalent carbon to a more stable tetravalent state, obscures the limited availability of isocyanides; only
24 isocyanides are commercially available at prices less than $50/g of which almost half are derivatives of Tosmic.
5 Structurally complex isocyanides therefore require assembly from simpler precursors, most often via sequential formylation and dehydration of primary amines.
6Asmic (
2),
Anisylsulfanylmethylisocyanide,
7 fills the void by providing an efficient
isocyanide building block that allows rapid access to structurally diverse substituted isocyanides
8 and heterocycles.
9 Formed by a sequential Mannich formylation-dehydration sequence,
Asmic is a bench-stable, crystalline solid that stores well with minimal odor.
Asmic is readily deprotonated by a variety of bases (NaH, LDA, BuLi) to afford a nucleophilic organometallic that efficiently intercepts electrophiles (Scheme 1,
2 →
3). The deprotonation-alkylation can be performed in separate steps or efficiently telescoped into a single synthetic operation to convert
Asmic into dialkylAsmic
3; the telescoped alkylation with α,ω-dihalides affords cyclic dialkylAsmic derivatives
3.
Treatment of dialkylAsmic 3 with BuLi triggers the rapid formation of a highly nucleophilic organolithium whose trapping with a variety of electrophiles efficiently generates tri-substituted isocyanides 4 (Scheme 1). The conversion of disubstituted Asmic derivatives 3 to reactive intermediates is extremely fast; the reaction with BuLi is essentially complete within 5 min at -78 °C. Subsequent trapping with an array of electrophiles ranging from alkyl halides to carbonyls to heteroatom species affords the corresponding trisubstituted Isocyanide (Scheme 1 4a - 4b, 4e - 4k, and 4n, respectively); ketones react to afford oxazolines formed by attack of the intermediate alkoxide on the electrophilic Isocyanide (Scheme 1, 4l and 4m). Disubstituted isocyanides are accessed through two sequential alkylations of Asmic followed by the addition of BuLi and NH4Cl (see 4c and 4d, Scheme 1). The Asmic-based syntheses allows an efficient route to substituted isocyanides with functionality that is otherwise difficult to access, such as the ketoisocyanides 4g and 4h.
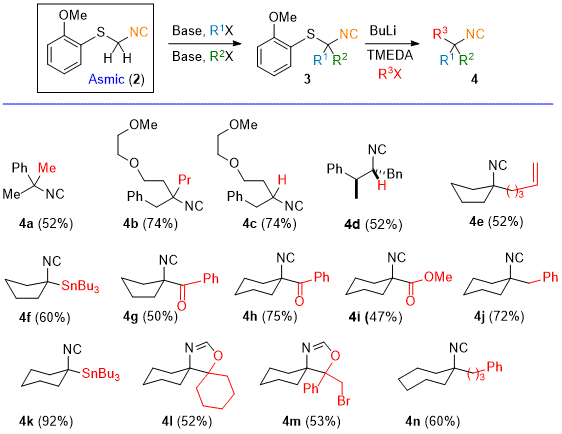
Scheme 1. Asmic-Based Synthesis of Di- and Tri-Substituted Isocyanides
DialkylAsmic derivatives
3 provide an efficient route to quaternary nitriles
6 (Scheme 2).
10 Omitting TMEDA during the addition of BuLi triggers a facile sulfur-lithium exchange-isomerization to afford the corresponding lithiated nitrile
5, excellent nucleophiles that efficiently intercept a range of electrophiles.
11 Trapping the lithiated nitriles
5 with carbon electrophiles installs quaternary centers. The BuLi-initiated isocyanide-nitrile isomerization is equally efficient in forming cyclic nitriles (
6a and
6b) as for acyclic nitriles (
6c and
6d).
Scheme 2. Asmic-Based Synthesis of Quaternary Nitriles
Asmic is a versatile isocyanide building block whose sequential alkylations afford di- and tri-substituted isocyanides. The sequenced double alkylation, BuLi-exchange-alkylation provides a rapid route to isocyanides or nitriles that are otherwise difficult to access. The Asmic-based isocyanide synthesis is ideally suited to accessing homologous isocyanides for structure activity assays because the strategy is modular in nature and highly efficient.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
N-((2-Methoxyphenylthio)methyl)formamide: Formamide, N-[[(2-methoxyphenyl)thio]methyl]-; (1) (118617-57-5)
Asmic, anisylsulfanylmethylisocyanide: Benzene, 1-[(isocyanomethyl)thio]-2-methoxy-; (2) (1803329-89-6)
Paraformaldehyde; (30525-89-4)
Formamide; (75-12-7)
Methoxybenzenethiol: (7217-59-6)
Diisopropylethylamine: N-Ethyl-N-(propan-2-yl)propan-2-amine; (7087-68-5)

|
Fraser Fleming obtained a BS (Hons.) at Massey University, New Zealand, in 1986 and a Ph. D. in 1990 from the University of British Columbia, Canada under the direction of Edward Piers. He completed postdoctoral research with James D. White at Oregon State University before taking his first faculty position at Duquesne University, Pittsburgh in 1992. In 2013 he began a two-year appointment as a Program Director at the National Science Foundation working in the Synthesis and the Catalysis Programs. In 2015 he moved to Drexel University where his research is focused on the reactions of isocyanides and nitriles. |

|
Embarek Alwedi received his B.S in 2001 and M.S in 2006 from Alfatah University, Libya. He remained at his alma mater for a further two years working as a lecturer before relocating to the US to pursue Ph.D. with Prof. Paul Blakemore at Oregon State University. He completed his postdoctoral studies under the supervision of Prof. Fraser Fleming before moving to Merck, Rahway where he is working as research chemist in process R&D. His research interests lie in the development of new synthetic methodology and processes. |

|
Bilal Altundas obtained his BS with honors in chemistry from Middle East Technical University in Ankara, Turkey in 2013 before moving to Miami University to pursue an MS in organic chemistry under the direction of Professor Hong Wang. Upon completing his MS in 2016, he started his Ph. D. under the guidance of Professor Fraser F. Fleming in the Department of Chemistry at Drexel University. His research interests are in synthetic methodology as applied to the reactions of ketenimines and in uncovering new reactions of metalated isocyanides. |

|
Allen Chao obtained a B.S. in chemistry from the University of Pittsburgh in 2007 followed by industrial experience before embarking on a Ph. D. He began his graduate career at Duquesne University, then moved with Prof. Fleming to complete his Ph. D. at Drexel University in 2018. He is currently working as a post-doctoral fellow in the Wistar Institute, Philadelphia. His research interests include development of PROTACs as therapeutic agents in treating cancer. He is invested in helping bridge the communication gap between the chemistry and biology disciplines. |

|
Zachary L. Ziminsky began his undergraduate career at Drexel University. He worked on the large-scale synthesis of Asmic as a summer research student with Prof. Fleming. |

|
Maanasa Natrajan completed her high school at Jnanodaya school, Bengaluru in India. In 2016, she started her undergraduate at Drexel University where she is currently a senior pursuing an individualized course of interdisciplinary study drawing upon neuroscience, chemistry, biophysics, and computation. She started research in organic chemistry with Prof. Fleming in her first year and then continued as a research assistant. She has performed systems biology research at Thomas Jefferson University, bioinformatics and phage biology at the Genome Institute of Singapore, and theoretical and computational neuroscience at Howard Hughes Medical Institute, Janelia. She plans to pursue a Ph. D. in neuroscience. |

|
Martina Drescher is the lead technician of the Maulide group at the University of Vienna, where she has worked with several group leaders over the course of 38 years. |

|
Anthony J. Fernandes received his Ph.D. in 2018 at the University of Bordeaux, under the supervision of Prof. Yannick Landais and Dr. Frédéric Robert. He moved to the University of Strasbourg and the University of Poitiers, starting a post-doctoral position within a collaboration with Dr. Frédéric Leroux and Prof. Sébastien Thibaudeau. He then moved to the University of Vienna, where he is currently working as a postdoctoral researcher in the group of Prof. Nuno Maulide. Research interests involve highly reactive intermediates, rearrangements and the synthesis of biologically active compounds. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved