Org. Synth. 2021, 98, 194-226
DOI: 10.15227/orgsyn.098.0194
Enantioselective Michael-Proton Transfer-Lactamization for Pyroglutamic Acid Derivatives: Synthesis of Dimethyl-(S,E)-5-oxo-3-styryl-1-tosylpyrrolidine-2,2-dicarboxylate
Christian M. Chaheine, Conner J. Song, Paul T. Gladen, and Daniel Romo*
1
Checked by Matthew J. Genzink and Tehshik P. Yoon
1. Procedure (Note 1)
A. Dimethyl 2-((4-methylphenyl)sulfonamido)malonate (2). To a single-necked, 500 mL round-bottomed flask containing a football-shaped, teflon-coated stir bar (5 cm) and fitted with a 24/40 glass, threaded gas-inlet adapter with a silicone/PTFE septa (Figure 1A) (Note 2), in-turn connected via chemically resistant tubing to a vacuum/nitrogen manifold is added commercially available dimethyl aminomalonate hydrochloride (1, 12.5 g, 68.2 mmol, 1.00 equiv) and p-toluenesulfonic anhydride (27.2 g of 90 wt. % purity, 74.9 mmol, 1.1 equiv) (Note 3) as solids. The atmosphere in the flask is replaced with nitrogen by three cycles of vacuum (Note 4) and nitrogen back-filling via the vacuum/nitrogen manifold. The glass adapter is briefly removed, and tetrahydrofuran (270 mL) is then added from a polyethylene graduated cylinder. The resulting beige suspension is stirred vigorously and cooled to 0 °C in an ice/water bath (e.g. recrystallizing dish, Figure 1B) for 15 min before adding freshly distilled triethylamine (28.5 mL, 204 mmol, 3.0 equiv; Note 5) by rapid dropwise addition via a 60 mL, plastic, luer-lock syringe fitted with a stainless steel 18-gauge needle over ~5 min. A yellow-orange suspension forms within 1 h (Figure 1C) and gradually becomes a light-yellow color as the reaction mixture is stirred over 24 h (Note 6) (Figure 1D) and allowed to slowly warm to ambient temperature (23 °C).

Figure 1. N-Tosylation of dimethylaminomalonate hydrochloride: A) Threaded gas-inlet adapter, 24/40 joint size, fitted with a red/white PTFE/silicone septa in an open-top screw cap. B) Prior to addition of triethylamine. C) 1 hour after addition of triethylamine. D) 16 h after addition of triethylamine (photos provided by submitters).
The yellow-orange suspension is then vacuum-filtered through a pad of celite (Note 7) (Figure 2A) into a 1 L, heavy-walled vacuum Erlenmeyer flask to remove the white precipitate by-product, which is triethylammonium p-toluenesulfonate. Quantitative transfer of the crude material to the filter funnel is carried out by rinsing with ethyl acetate from a polyethylene squirt bottle (~25 mL). The filter cake is rinsed with ethyl acetate (250 mL) and the filtrate is transferred to a 1 L recovery flask and concentrated by rotary evaporation (~150 to 75 mmHg, 30 °C bath temperature) to a yellow oil that is dry-loaded onto 70 g of silica gel (Notes 8 and 9). The resulting fine yellow powder is loaded onto a pre-packed flash chromatography column for purification (Figures 2B-C) (Note 10) and eluted to yield 13.3 g (65%) of dimethyl 2-((4-methylphenyl)sulfonamido)malonate (2) as an off-white grainy powder (Note 11) (Figure 2D).

Figure 2. N-Tosylation reaction work-up and column chromatography: A) Crude reaction solution after filtering through Celite. B) Flash column prior to fraction collection with crude mixture pre-adsorbed onto silica gel. C) Flash column following product collection; yellow color that progresses through column is not collected. D) Appearance of N-tosyl-2-aminomalonate (photos provided by submitters).
B. (2E,4E)-5-Phenylpenta-2,4-dienoyl chloride (4). To an oven-dried (Note 12), 500 mL, single-necked, round-bottomed flask containing a football-shaped, teflon-coated stir bar (4 cm) and fitted with a 24/40 glass, threaded gas-inlet adapter with a silicone/PTFE septa (Figure 1A, Note 2), in-turn attached via chemically resistant tubing to a vacuum/nitrogen manifold, is added commercially available 5-phenyl-2,4-pentadienoic acid (3) (10.0 g, 57.5 mmol, 1.00 equiv) (Note 13). The atmosphere in the vessel is replaced with nitrogen by three cycles of evacuation (Note 4) and back-filling with nitrogen via the vacuum/nitrogen manifold. The hose connection to the manifold is then quickly replaced with tygon tubing connected in sequence to a mineral oil bubbler followed by a solution of saturated, aqueous sodium bicarbonate (150 mL) in a 250 mL Erlenmeyer flask (Figure 3A). Dichloromethane (230 mL) is then added in four portions via a 60 mL plastic syringe fitted with an 18-gauge stainless steel needle, forming a beige-colored suspension that is stirred at ambient temperature (23 °C). N,N-Dimethylformamide (0.67 mL, 8.6 mmol, 15 mol%) is added via a plastic 1 mL syringe in one portion, followed by dropwise addition of oxalyl chloride (14.6 mL, 173 mmol, 3.0 equiv) over 15 min via a 30 mL plastic luer-lock syringe fitted with a 20-gauge stainless-steel needle connected to a syringe pump (59 mL/h flow rate). The reaction mixture is stirred at ambient temperature (23 °C) while venting gaseous by-products through the mineral oil bubbler/sodium bicarbonate solution (Note 14) until it became a homogenous, gold-colored solution and had ceased gas evolution (~3-4 h).

Figure 3. Acid chloride synthesis: A) Reaction set up with mineral oil bubbler and sodium bicarbonate solution in sequence to control and quench exhaust gases. B) Crude acid chloride used in the next reaction after drying on high vacuum with an attached acid scrubber (KOH pellets) (photos provided by submitters).
The stir bar is removed and the crude reaction mixture is concentrated by rotary evaporation (150 to 75 mmHg, 30 °C bath temperature, ~1 h) to a light-brown, amorphous solid that is further dried under high vacuum (~3 mmHg) (Note 15) (Figure 3B) with an intervening acid scrubber column for at least 3 h to provide (2E,4E)-5-Phenylpenta-2,4-dienoyl chloride (4). The atmosphere in the flask is charged with nitrogen via the Schlenk manifold yielding 11.2 g (101%) of crude acid chloride as a yellow solid which is used directly in the next reaction without purification (Note 16).
C. Dimethyl-(S,E)-5-oxo-3-styryl-1-tosylpyrrolidine-2,2-dicarboxylate (5): To an oven-dried (Note 12), 1 L, single-necked, round-bottomed flask containing a football-shaped, teflon-coated stir bar (5 cm) is added dimethyl sulfonamidomalonate (2) (12.4 g, 41.3 mmol, 1.00 equiv). The reaction vessel is fitted with a T-bore Schlenk adapter attached to both vacuum and an argon balloon (Notes 17 and 18) (Figure 4A). The atmosphere in the flask is replaced with argon by three cycles of vacuum/back-filling via the Schlenk adapter, which is then quickly replaced with a rubber septum and an argon balloon. Tetrahydrofuran is added (210 mL, 0.20 M initial concentration of sulfonamidomalonate) (Notes 19 and 20) in four portions via a plastic 60 mL syringe fitted with an 18-gauge stainless steel needle. The resulting clear solution is stirred vigorously and cooled to 0 °C in an ice bath over 15 min. A newly opened 100 mL bottle of lithium hexamethyldisilazide (LiHMDS, 1.0 M in tetrahydrofuran, 47.5 mL, 47.5 mmol, 1.15 equiv) (Note 21) is then added dropwise from a 60 mL plastic luer-lock syringe fitted with an 18-gauge stainless-steel needle connected to a syringe pump over 15 min (189 mL/h flow rate, Figure 4B).

Figure 4. Nucleophile-catalyzed Michael-proton transfer-lactamization (NCMPL); Deprotonation of sulfonamidomalonate: A) T-bore Schlenk adapter attached to 1 L, single-necked round-bottomed flask. B) Addition of LiHMDS solution via syringe pump. C) Following complete addition of LiHMDS (photos provided by submitters).
The resulting yellow solution (Figure 4C) is further cooled for 15 min in a cryobath set to -10 °C (Note 22). 1,8-Diazobyciclo[5.4.0]undec-7-ene (DBU, 6.85 mL, 45.4 mmol, 1.10 equiv) is then added as a very viscous liquid in a single portion via a 12 mL plastic syringe equipped with an 18 gauge needle followed by the addition of O-trimethylsilylquinine (TMSQN) solution via a 12 mL plastic syringe via a 20 gauge needle in one portion (Note 23) within 5 minutes of DBU addition. A solution of crude, (2E,4E)-5-Phenylpenta-2,4-dienoyl chloride (4) (1.0 M in tetrahydrofuran, 57.5 mmol, 1.39 equiv in 57.4 mL) (Note 24) is then added dropwise over 3 h from a 60 mL luer-lock plastic syringe fitted with a 16-gauge stainless-steel needle connected to a syringe pump (19 mL/h flow rate; Figure 5B-C).

Figure 5. NCMPL Organocascade: A) Appearance of crude unsaturated acid chloride solution in THF. B) Reaction solution following addition of DBU and TMSQN. C) Addition of crude unsaturated acid chloride solution via syringe pump with cooling from a cryobath system (photos provided by submitters).
The reaction mixture is stirred at -10 to -14 °C for an additional 3 h (total reaction time = 6 h) (Note 25) and is quenched by carefully pouring the cold orange-red solution into a 1 L separatory funnel containing aqueous hydrochloric acid (1M, 150 mL). Quantitative transfer of the crude reaction mixture to the separatory funnel is carried out by rinsing the reaction vessel with ethyl acetate (150 mL). The layers are separated (Figure 6A) and the aqueous layer is extracted with ethyl acetate (4 x 125 mL) (Figure 6B). The combined organic layers are washed with saturated, aqueous sodium chloride solution (150 mL), dried over magnesium sulfate (5 g), and filtered through Celite (Note 7) (Figure 6C) into a 1 L Erlenmeyer flask. The filter cake is rinsed with ethyl acetate (75 mL).

Figure 6. NCMPL reaction work up: A) Formation of layers in separation funnel upon first extraction with ethyl acetate. B) Appearance of layers in separation funnel following final extraction with ethyl acetate. C) Combined organic layers after drying and filtering through Celite (photos provided by submitters).
The filtrate is transferred to a 2 L recovery flask and concentrated by rotary evaporation (~200 mm Hg, 30 °C bath temperature) until a yellow, amorphous solid forms (Figure 7A). This crude, yellow solid is transferred to a Büchner funnel (7 cm diameter) by use of a stainless-steel spatula. The solid is agitated with a spatula and washed with a minimal amount of methanol (~200 mL) from a polyethylene squirt bottle while under vacuum filtration (Note 26) (Figure 7B). After transferring the off-white solid to 6-dram vials and removing the residual solvent under high vacuum for at least 4 h (Notes 4 and 27), 13.5 g of Dimethyl-(S,E)-5-oxo-3-styryl-1-tosylpyrrolidine-2,2-dicarboxylate (5) (71% isolated yield, >99:1 er) is obtained as an off-white crystalline powder that is of sufficient purity (97% by q1H-NMR) for subsequent reactions (Notes 28 and 29).

Figure 7. NCMPL product isolation: A) Crude solid following thorough removal of volatiles by rotary evaporation. B) Product after washing with methanol under gentle vacuum filtration in a Büchner funnel. C) Appearance of γ-lactam (5) as a powdery crystalline solid (photos provided by submitters).
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at https://www.nap.edu/catalog/12654/prudent-practices-in-thelaboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
dimethyl aminomalonate hydrochloride,
p-toluenesulfonic anhydride,
triethylamine,
5-phenyl-2,4-pentadienoic acid,
oxalyl chloride,
methylene chloride,
N,N-dimethylformamide, mineral oil,
sodium bicarbonate,
dihydrogen monoxide,
carbon monoxide,
carbon dioxide,
hydrogen chloride,
lithium hexamethyldisilazide,
1,8-diazobyciclo[5.4.0]undec-7-ene,
methanol,
tetrahydrofuran,
ethyl acetate,
hexanes,
Celite,
magnesium sulfate, and silica gel.
2. A rubber septum and needle connected to a nitrogen/vacuum manifold can also be used if the glass adapter shown in Figure 1A is unavailable.
3.
Dimethyl aminomalonate hydrochloride (97%) was purchased from Sigma Aldrich,
p-toluenesulfonic anhydride (90%) was purchased from Oakwood,
triethylamine (99%),
tetrahydrofuran (HPLC grade, unstabilized)
ethyl acetate (ACS grade), and
hexanes (ACS grade) were purchased from Fisher Scientific. All reagents and solvents, excluding
triethylamine, were used as received.
p-Toluenesulfonyl chloride (
TsCl) was also studied for this tosylation but was ineffective even in the presence of
DMAP.
4. A Welch Duo Seal 1400 rotary vane vacuum pump (~3 mm Hg) connected to the vacuum/nitrogen manifold through a cold-trap was used for high-vacuum applications throughout this procedure.
5.
Triethylamine was distilled from calcium hydride (95%, purchased from Oakwood) under an atmosphere of dry nitrogen prior to use.
6. The formation of product can be monitored by thin-layer chromatography (TLC, 30%
ethyl acetate/
hexanes, sulfonamidomalonate product R
f = 0.40, visualized under a 254 nm UV-lamp). Glass-backed, 250 μm-thickness TLC plates were purchased from Silicycle inc.
Figure 8. TLC analysis of Step A (photo provided by submitters)
7.
Celite 545 filter aid was purchased from Fisher Scientific. A pad of
Celite (1.5 cm tall) was used in a 150 mL funnel (6.5 cm height x 7 cm inner diameter) with a glass frit of porosity M.
8. Silica Gel (Silicycle Ultrapure SilicaFlash silica gel, 60 Å pore size, particle size distribution 40-63 microns) was purchased from Fisher Scientific.
9. Silica gel (70g) was added to the crude yellow oil following rotary evaporation.
Ethyl acetate was used to rinse the walls of the 1 L recovery flask to ensure complete and uniform absorption onto the silica. Solvent that could interfere with chromatographic separation was thoroughly removed by rotary evaporation until the yellow powder thus formed was dry and free-flowing.
10. A heavy-walled, glass flash chromatography column 10 cm in diameter was used. The free-flowing fine yellow powder silica gel with the absorbed crude material was carefully loaded into the column, which, beforehand, was wet-packed with 250 g of fresh silica gel using
hexanes and topped with a layer of sand. A step-wise gradient was used (
hexanes (500 mL), then 20%
ethyl acetate/
hexanes (500 mL), then 40%
ethyl acetate/
hexanes (500 mL), then 60%
ethyl acetate/
hexanes (2500 mL). The eluent received when eluting with
hexanes was collected in a 1 L Erlenmeyer flask. The eluent from 20%
ethyl acetate in
hexanes was collected in 500 mL Erlenmeyer flasks. The product was then collected in 250 mL fractions using Erlenmeyer flasks starting at 40% and ending at 60%
ethyl acetate/
hexanes. Product elution was monitored via thin-layer chromatography (50%
ethyl acetate/
hexanes, R
f = 0.60 visualized under a 254 nm UV-lamp). Glass-backed 250 μm-thickness TLC plates were purchased from Silicycle, Inc.

Figure 9. TLC analysis of column fractions for Step A (photo provided by submitters)
11. Characterization data for
dimethyl 2-((4-methylphenyl)sulfonamido)-malonate (
2): IR (thin film) 3235, 1749, 1160 cm
-1;
1H NMR
pdf (500 MHz, CDCl
3) δ: 2.43 (s, 2H), 3.68 (s, 3H), 4.69 (d,
J = 8.5 Hz, 1H), 5.56 (d,
J = 8.5 Hz, 1H), 7.30 (d,
J = 8.3 Hz, 1H), 7.74 (d,
J = 8.3 Hz, 1H);
13C NMR
pdf (125 MHz, CDCl
3) δ: 21.6, 53.6, 58.5, 127.3, 129.7, 136.4, 144.1, 166.0; mp = 118-122 °C; HRMS (ESI) [M + H]
+ calculated for C
12H
15NO
6S: 302.06928, found: 302.0686; This compound can be stored at ambient temperature (23 °C) indefinitely without decomposition. The purity was assessed at 82% by qNMR
pdf using 14.2 mg of the product and dimethyl terephthalate (20.2 mg) as an internal standard. The corrected yield for the reaction would be 10.9 g (53%). The checkers performed a second reaction yielding 13.9 g (69%) of the product.
12. Reaction flasks were dried in an oven at 125 °C for at least 4 h prior to use.
13.
5-Phenyl-2,4-pentadienoic acid (98%), purchased from Combi-Blocks,
oxalyl chloride (99% reagent plus), purchased from Sigma Aldrich, and
N,N-dimethylformamide (99.8% extra dry, AcroSeal), purchased from Acros, were used as received.
Methylene chloride (HPLC grade, cyclohexane preservative) was purchased from Fisher Scientific and passed through a column of activated alumina under an atmosphere of ultra-high purity argon (JC Meyer Solvent Purification System) prior to use. It was empirically determined that 3.0 equiv of
oxalyl chloride was required for complete consumption of the starting carboxylic acid.
14.
Caution! This reaction rapidly expels gaseous by-products:
carbon dioxide, caustic
hydrogen chloride, and toxic
carbon monoxide. Thus, careful dropwise addition of neat
oxalyl chloride is performed in a uniform, controlled fashion using a syringe pump over ~15 min.
15. A column of
potassium hydroxide pellets (Fisher Scientific) was connected in-line between the Schlenk manifold and the round-bottomed flask with a fritted gas outlet adapter and thick-walled Tygon tubing to act as an acid scrubber and prevent
HCl corrosion of the vacuum pump.
16. This compound (
4) could be stored for up to a week under inert atmosphere in a sealed vessel at -20 °C, but both the submitters and checkers used it immediately. The checkers performed a second reaction on half scale yielding 5.64 g (102%) of the crude product.
17. The submitters found the use of an argon balloon was more convenient in providing an inert atmosphere for the nucleophile-catalyzed Michael-proton transfer lactamization, but a typical nitrogen line with a needle and rubber septum can also be used.
18. Thick-walled, natural latex rubber balloons were purchased from Sigma Aldrich (SKU- Z154997).
19.
Tetrahydrofuran (HPLC grade, unstabilized) was purchased from Fisher Scientific and passed through a column of activated alumina under an atmosphere of ultra-high purity argon (JC Meyer Solvent Purification System).
Methanol (ACS grade) was purchased from Fisher Scientific and used as received.
Lithium hexamethyldisilazide (1.0 M in
tetrahydrofuran) and
1,8-diazobyciclo[5.4.0]undec-7-ene (
DBU, 98%) were purchased from Sigma Aldrich, and both were used as received. Sodium chloride was purchased from Fisher Scientific and used as received.
Magnesium sulfate (99%, anhydrous powder) was purchased from Oakwood and used as received by the submitters.
Magnesium sulfate (97%, anhydrous powder) was purchased from Sigma Aldrich and used as received by the checkers.
20. The initial concentration of the sulfonamidomalonate starting material is essential to the yield of this reaction as variations led to formation of insoluble salts as the base is added and greatly reduced yields.
21. The quality of the
LiHMDS used is very important and a newly opened 100 mL bottle for this procedure is required to ensure titer of base is as close as possible to the nominal 1.0 M indicated. The appearance of the
LiHMDS solution in the plastic syringe should be a very light yellow/orange in color and clear (not dark orange or cloudy). Complete formation of the enolate from the starting sulfonamidomalonate is essential to the yield of the product lactam.
22. Maintaining the reaction temperature between -10 and -14 °C is critical to obtain high enantioselectivity in the initial Michael-addition step.
23.
O-Trimethylsilylquinine (
TMSQN), an off-white, amorphous solid, was prepared according to the
Organic Syntheses procedure previously reported by the submitters.
2 TMSQN is prepared as a solution in
tetrahydrofuran under an atmosphere of nitrogen (1.0 M, 3.29 g, 8.30 mmol, 0.20 equiv in 8 mL
THF) in an oven-dried (
Note 12) 25 mL pear-shaped flask fitted with a glass, threaded gas-inlet adapter with a silicone/PTFE septa (
Note 2).
24. Both the submitters and checkers observed a light-yellow, undissolved solid which remained undissolved when forming the
THF solution of the crude unsaturated acyl chloride. While some solid may be pulled into the syringe prior to dropwise addition to the reaction mixture, it does not create an issue with the reaction. However, the syringe/syringe pump apparatus should be checked periodically to ensure the needle is not clogged and regular dropwise addition is proceeding.
25. The consumption of starting material and the formation of the product could be monitored by thin-layer chromatography (TLC, 30%
ethyl acetate/
hexanes, sulfonamidomalonate substrate R
f = 0.40, lactam product R
f = 0.50, visualized under a 254 nm UV-lamp) Glass-backed, 250 μm-thickness TLC plates were purchased from Silicycle, Inc.
Figure 10. TLC analysis of Step C (photo provided by submitters)
26. The
methanol rinse under vacuum filtration removes the yellow color of the crude solid and should be done with caution to prevent the resulting fine crystalline powder from passing underneath the filter paper. The product is sparingly soluble in MeOH. Up to 200 mL of
methanol can be required to completely remove the yellow impurity. A stainless steel spatula is used to break apart clumps of yellow solid to expose product surface for thorough rinsing.
27. Thorough removal of residual
methanol from the product by high vacuum is necessary in order to avoid decomposition upon storage at ambient temperature.
28. Characterization data for
dimethyl-(S,E)-5-oxo-3-styryl-1-tosylpyrrolidine-2,2-dicarboxylate (
5): Absolute stereochemistry was assigned by the submitters based on X-ray analysis using anomalous dispersion; [α]
20D +0.78 (
c 10.0, CHCl
3); IR (thin film) 1736, 1162 cm
-1; mp = 164-165 °C;
1H NMR
pdf (500 MHz, CDCl
3) δ:2.45 (s, 3H), 2.65 (dd,
J = 16.9, 9.4 Hz, 1H), 2.70 (dd,
J = 17.0, 8.3 Hz, 1H), 3.54 (app q,
J = 8.5 Hz, 1H), 3.81 (s, 3H), 3.94 (s, 3H), 6.07 (dd,
J = 15.8, 8.0 Hz, 1H), 6.47 (d,
J = 15.8 Hz, 1H), 7.36-7.27 (m, 7H), 8.06 (d,
J = 8.4 Hz, 2H);
13C NMR
pdf (125 MHz, CDCl
3) δ: 21.7, 35.8, 45.1, 53.4, 53.8, 76.2, 123.3, 126.6, 128.4, 128.7, 129.0, 129.9, 134.5, 135.1, 135.8, 145.4, 165.9, 167.5, 171.6; HRMS (ESI) [M + Na]
+ m/z calculated for C
23H
23NO
7S: 480.1087, found: 480.1077; This compound can be stored at ambient temperature (23 °C) on the benchtop indefinitely without decomposition. The purity was assessed at 97% by qNMR
pdf using 12.1 mg of the product and dimethyl terephthalate (22.7 mg) as an internal standard. The corrected yield as per qNMR
pdf would be 13.1 g (69%). The checkers performed a second reaction on half scale yielding 6.4 g (67%) of the product.
29. The submitters determined the enantiomeric ratio by chiral-HPLC analysis in comparison with an authentic racemic sample (synthesized using racemic catalyst (
i.e. 1:1
TMSQN/
TMSQD)) of the product using a Chiralcel AD-H column:
hexanes/
iPrOH = 80:20, flow rate of 0.5 mL/min, λ = 254 nm: t
minor = 75.3 min, t
major = 80.1 min. The checkers determined the enantiomeric ratio by chiral-SFC analysis in comparison with an authentic racemic sample (synthesized using racemic catalyst (
i.e. 1:1
TMSQN/
TMSQD)) of the product using a Chiralcel OJ-H column: 10%
iPrOH, flow rate of 3 mL /min, t
major = 11.0 min, t
minor = 14.8 min.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The development of synthetic methods for enantioselective access to functionalized pyroglutamate,
3, 4 and, more generally, γ-lactam (aka pyrrolidine-2-one, γ-butyrolactam, azolidine-2-one, 2-oxopyrrolidine) containing compounds remains a highly active topic in the field of organic synthesis. This is due to their versatility as synthetic intermediates and their presence in many natural products, metabolites, and pharmaceuticals with broad-ranging and potent biological activities (Figure 11).
5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22Figure 11. Enantioselective transition metal-catalyzed γ-lactam synthesis
There are an abundance of chemical methods for the synthesis of γ-lactams in racemic form, primarily through intramolecular cyclizations of functionalized linear substrates via C-N as well as C-C bond formation.
7,21,23,24,25,26,27 In only the last three years, several methods for the construction of racemic γ-lactams have been reported- tandem addition/cyclization,
28,29,30,31 multi-component,
32,33,34,35 intramolecular C-H activation
36,37,38,39,40, photoredox-catalyzed,
41,42,43 transition metal-catalyzed cyclizations,
44,45 rearrangement,
46 direct α-alkylation of primary amines with acrylates,
47 ring contraction/expansion,
48,49 and flow chemistry.
50 Chemoenzymatic,
51 enzymatic resolution,
52 and chiral pool synthesis
53,54 have also been reported recently as asymmetric strategies. While these myriad, powerful methods are now available for the synthesis of γ-lactams, fewer examples illustrate a robust means of
enantioselective construction of this key
N-heterocycle from achiral starting materials. Even fewer methods have demonstrated scalability while maintaining a high degree of enantiopurity in the product.
Enantioselective synthesis of γ-lactams:
The following will briefly highlight recent (within the last three years) advances in synthetic methods to access chiral γ-lactams in an enantioselective fashion from achiral starting materials. This will be followed by a discussion of our group's research activity in the area of unsaturated acylammonium catalysis, centering on the nucleophile-catalyzed Michael-proton transfer lactamization (NCMPL) used in the described procedure.
Asymmetric organocatalytic routes to γ-lactams primarily employ chiral
N-heterocyclic carbenes (NHCs),
55 which is a complementary strategy to the NCMPL methodology in its use of α,β-unsaturated carbonyl derivatives as substrates in a cascade process. In addition to multiple NHC-based organocatalytic methods, chiral phosphoric acids,
56 phase-transfer,
57 and hydrogen bond donor catalysts,
58 and multi-component reactions
59,60 have also appeared recently as alternate avenues to optically active γ-lactams. Huang disclosed a Michael-addition/cyclization reaction using chiral NHC
A3 as a precatalyst, α-bromo-α,β-enals and α-sulfonamidoketones as substrates, and an excess of alkylamine base (Scheme 1A).
61 High-enantioselectivities and diastereoselectivites are obtained, and the reaction was performed on gram scale without significant loss of yield or enantiopurity. Notably, heteroaryl and cycloalkyl substituted ketones are efficient reactants in this process, delivering functionalized
trans-γ-lactams that are aptly suited to the synthesis of clausenamide analogues.
A chiral NHC-catalyzed homoenolate addition/cyclization was reported by Li.
62 Utilizing the same precatalyst
A3 and various α,β-enals as homoenolate precursors, the Li group demonstrated a formal (3+2) annulation occurs with α-sulfonamidoacrylates (Scheme 1B). Mechanistic investigations revealed that, rather than the anticipated homoenolate Michael-addition/cyclization sequence, a tautomerization of the sulfonamidoacrylate to an α-iminoester precedes homoenolate addition and cyclization, yielding pyroglutamate derivatives that bear an
aza-quaternary center in good yield and enantioselectivity.
Scheme 1. Organocatalytic enantioselective γ-lactam synthesis
In contrast, Córdova developed an asymmetric cascade processes to construct bicyclic γ- and δ-lactam
N,S-acetals through chiral amine catalysis (Scheme 1C).
63 The bicyclic products are intriguing scaffolds that occur in 5-membered ring analogs of penicillin antibiotics. In this reaction, Mannich-addition of an enolizable aldehyde onto an α-iminoester is catalyzed by (
S)-proline, forming a Mannich-base intermediate that is then treated with an aminothiol to afford bicyclic α-amino as well as α-hydroxy-γ-lactam
N,S-acetals with excellent enantioselectivity.
More prevalent in the literature are methods based on transition metal catalysis. The asymmetric reductive amination of α-ketoesters for the synthesis of enantioenriched
N-unprotected-γ-lactams was realized by the Yin group.
64 A chiral bidentate phosphine ligand
L1 in combination with Ru(OAc)
2 under an atmosphere of H
2 was identified as an efficient catalytic system for the reductive amination to
NH-γ-lactams (Scheme 2A). Only aryl-substituted ketone substrates led to high enantioselectivities, however. This method was used on up to a gram-scale to build a pyrrolidine intermediate toward an anti-cancer therapeutic, Larotrectinib, as well as a benzolactam intermediate toward a quinolone antibiotic drug, Garenoxacin.
An enantioselective cobalt-catalyzed hydroboration cyclization of linear
N-allyl-propiolamides was recently reported by Ge.
65 Borylated γ-lactams bearing stereogenic all-carbon quaternary centers were constructed in high yield and enantioselectivity with the application of chiral bidentate phosphine ligand
L2 and Co(acac)
2 (Scheme 2B). This reaction was conducted on a gram-scale and the versatility of the boryl functionality was demonstrated through a variety of known transformations.
Scheme 2. Enantioselective transition metal-catalyzed γ-lactam synthesis
Activation of sp
3 C-H bonds by transition metal catalysis is an important and fertile sub-field in organometallic chemistry and recently has enabled the enantioselective synthesis of NH-γ-lactams from readily-available carboxylic acid derivatives like 1,4,2-dioxazol-5-ones.
66,67,68 Chang optimized the intramolecular, Ir (III)-catalyzed γ-C-H amidation using a chiral diamine ligand and 1,4,2-dioxazol-5-ones as acylnitrenoid precursors (Scheme 2C).
69 The Curtius rearrangement pathway that commonly impedes the desired C-H-amidation through the formation of isocyanate side-products is completely suppressed with Ir catalyst
A1 under their established conditions. An impressive substrate scope is demonstrated using a variety of aliphatic sp
3 C-H containing dioxazolones, leading to functionalized γ-stereogenic-γ-lactams in good yields and high enantioselectivities. The reaction can be carried out in air and has been demonstrated on half-gram scale without erosion of product enantiopurity.
Alkenyl C-H-activation has also been employed recently in the enantioselective synthesis of γ-lactams by Cramer.
70 Acrylamides and allenes are reacted in a (4+1) annulation to γ-stereogenic-α,β-unsaturated-γ-lactams via chiral cyclopentadienyl Rh (III) catalyst
A2 (Scheme 2D). AgOAc is used to generate the active Rh (III) catalyst by ligand substitution and Cu(OBz)
2 was found to be the optimal source of terminal oxidant to regenerate the catalyst. Mild heating and extended reaction times (2-4 days) lead to chiral α,β-unsaturated-γ-lactams in good yields and high enantioselectivities, although the scalability of this method is unclear. Other methods of note for the enantioselective construction and reactions of γ-lactams include hydrogenation,
71,72 C-H functionalization,
73,74 desaturation,
75 and cyclopropanations.
76Synthetic utility of α,β-unsaturated acylammonium salts:
Initial reports of acylammonium salts as reactive intermediates emerged as early as the 1930s with Wegler's description of an asymmetric acyl-transfer process.
77 The synthetic potential of acylammonium salts has since expanded to include a multitude of asymmetric organocascade processes initiated by different types of chiral acylammonium intermediates (Figure 12), leading to mono- and polycyclic frameworks of varying complexity. Developments in this area have been recently reviewed.
78,79 In the last four years additional examples of asymmetric organocascade reactions using α,β-unsaturated acylammonium salts have been reported.
Figure 12. Chiral acylammonium salt intermediates
In 2016, Birman disclosed an asymmetric thio-Michael addition-aldol-β-lactonization-decarboxylation cascade process toward the synthesis of thiochromenes and in a related process accessed thiochromanes (Scheme 3).
80 Catalyzed by HBTM-2.0
81 or H-PIP
82, highly enantioselective generation of these heterocycles starting from readily available α,β unsaturated thioesters was demonstrated.
Scheme 3. Asymmetric thio-Michael addition-aldol-β-lactonization-decarboxylation cascade and thio-Michael-Michael-enol lactonization
In the same year, Smith reported chemo- and enantioselective, benzazole annulations catalyzed by HBTM-2.1 (
A5) that employ α,β-unsaturated homoanhydride electrophiles and afford benzazole δ-lactams and lactones from acylbenzothiazole and acylbenzoxazole nucleophiles (Scheme 4).
83 Computational studies suggested that the presence of two 1,5-S-O n→σ* interactions following the initial Michael-addition step, dictate the chemoselectivity of δ-lactone vs δ-lactam formation.
Scheme 4. Chemo-and enantioselective benzazole annulation catalyzed by HBTM-2.1
Until recently, catalytic turnover of the chiral Lewis-base in acylammonium organocascades was driven by acyl substitution with a pendant,
in situ generated nucleophile. In 2017, Smith showed that catalyst regeneration can be facilitated by an aryloxide counterion that is produced following the formation of the acylammonium intermediate from α,β-unsaturated aryl esters, expanding the scope of potential nucleophiles applicable in these types of organocascades (Scheme 5).
84 The Smith group demonstrated the utility of this approach with an enantioselective isothiourea-catalyzed Michael-addition of nitroalkanes to the α,β-unsaturated aryl esters in up to 79% yield and 99:1
er. The branched nitroalkene products could be elaborated to optically active γ-lactams following chemoselective reduction of the nitro moiety. Smith quite recently described the use of aryloxide-facilitated turnover toward the enantioselective Michael-addition of
N-heterocyclic pronucleophiles
85 and silylnitronates
86 to α,β-unsaturated aryl esters.
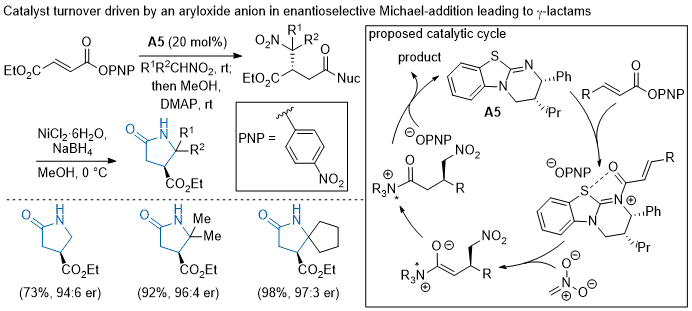
Scheme 5. Catalyst regeneration facilitated by an aryloxide counterion produced following acylammonium formation
A long-standing research focus of our group has been the application of chiral acylammonium salts in the organocatalytic, asymmetric synthesis of heterocycles. Since 2001, we developed a nucleophile-catalyzed/aldol/lactonization (NCAL),
87,88 nucleophile-catalyzed Michael/aldol/β-lactonization (NCMAL),
89 Diels-Alder/lactonization (DAL),
90,91 and a nucleophile-catalyzed Michael/proton transfer/lactamization (NCMPL),
92 the basis of the current procedure.
Nucleophile-catalyzed Michael/proton transfer/lactamization (NCMPL)
In 2013, we disclosed an enantioselective, organocatalytic nucleophile-catalyzed Michael/proton transfer/lactamization (NCMPL) cascade. Using α,β-unsaturated acylammonium salts generated from commercially available α,β-unsaturated acid chlorides, readily accessible O-trimethylsilylquinidine (TMSQD) or O-trimethylsilylquinine catalysts (TMSQN, A6), and N-protected aminomalonates as bis-nucleophilic reaction partners. The substrate scope from the original publication as well as the product of the current procedure (a new entry in the table) is illustrated below (Table 1).
During optimization of this method, the importance of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as an acid scavenger was noted, omitting this amine base resulting in <5% yield. The use of cinchona alkaloid derived, as opposed to isothiourea Lewis-bases, led to superior enantioselectivities. Additionally, substitution of LiHMDS with sodium bis(trimethylsilyl)amide (NaHMDS) also drastically reduced enantioselectivity, implicating the lithium cation as a crucial coordinating Lewis acid in the transition-state arrangement. This method was also applicable to the enantioselective synthesis of enol lactones when β-ketoesters are used as bis-nucleophilic reactants. The current procedure represents another example of the robustness of the NCMPL wherein a functionalized lactam is produced as virtually a single enantiomer on a decagram scale. The submitters have been able to scale the reaction up to 20 g of sulfonamidomalonate starting material without detriment to yield or enantiopurity. The above procedure can also be conducted using diethylaminomalonate hydrochloride, which is significantly less costly, however, the submitters were unable to find conditions for the recrystallization of the product γ-lactam from the crude reaction mixture. The exclusion of column chromatography following the NCMPL to obtain the desired γ-lactam product, as is possible in the case of dimethylaminomalonate, significantly simplifies the purification and was chosen for this reason.
Table 1. Substrate scope of NCMPL organocascade
Further expanding on the utility of chiral, α,β-unsaturated acylammonium salts, we adapted the NCMPL to the synthesis of medium-sized lactams (Table 2).
93 The resulting azepanones, benzazepinones, azocanones, and benzazocinones were generated in high enantiopurity and synthetically useful yield.
In summary, the NCMPL organocascade provides a convenient route to functionalized, small to medium-sized lactams, including
pyroglutamic acid derivatives, in optically active form typically with high enantiopurity. The utility of the derived lactams, including applications to natural product synthesis, and established manipulations can be found in the published work from our group in this area.
92, 93, 94
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Dimethyl aminomalonate hydrochloride; (1) (16115-80-3)
p-Toluenesulfonic anhydride; (4124-41-8)
THF: Tetrahydrofuran; (109-99-9)
Triethylamine: N,N-Diethylethanamine, (121-44-8)
5-Phenyl-2,4-pentadienoic acid; (3) (1552-94-9)
Dichloromethane; Methylene chloride; (75-09-2)
N,N-Dimethylformamide; (68-12-2)
Oxalyl chloride: Ethanedioyl dichloride; (79-37-8)
LiHMDS: Lithium hexamethyldisilazide; (4039-32-1)
DBU: 1,8-Diazobyciclo[5.4.0]undec-7-ene; (6674-22-2)

|
Christian Michael Chaheine was born in El Paso, Texas. He obtained his bachelor's degree in biochemistry from the University of Texas at Arlington in 2014 and began graduate studies in synthetic organic chemistry under the mentorship of Prof Daniel Romo at Texas A&M University in the same year. He moved with the Romo group to Baylor University in the fall of 2015 where his research efforts centered around the total synthesis of the oxazolomycins and simplified derivatives toward the elucidation of new bioactivities and protein target identification. He began post-doctoral studies with Prof. Christopher Parker (Scripps Florida) in 2021. |

|
Conner J. Song received his B.S. in Biochemistry & Molecular Biology and his B.A. in Art History from Wake Forest University in 2019 where he worked on characterizing prostate cancer stem cells under the mentorship of Dr. Bethany Kerr. He is currently working toward his Ph.D. under the guidance of Dr. Daniel Romo at Baylor University. His research is focused on mechanism of action studies of the curromycins and related natural products. |

|
Paul T. Gladen obtained a B.A. in Chemistry from St. Olaf College and a PhD in Organic Chemistry from Indiana University where he studied the synthesis of complex terpenoid natural products with Professor David Williams. He continued his study of natural products as a postdoctoral research associate at Texas A&M and Baylor University with Professor Daniel Romo before joining the faculty at Lake Forest College as an Assistant Professor in 2016. His independent research is in the area of natural product synthesis with a current focus on the synthesis of antimicrobial heterocycles. |

|
Daniel Romo received his B.A. in Chemistry/Biology from Texas A&M University and a Ph.D. in Chemistry from Colorado State University as a NSF Minority Graduate Fellow under the tutelage of the late Professor Albert I. Meyers. Following postdoctoral studies at Harvard as an American Cancer Society Fellow with Professor Stuart L. Schreiber, he began his independent career at Texas A&M in 1993 and then moved to Baylor University in 2015 where he is the Schotts Professor of Chemistry. Research interests in the Romo Group are at the interface of chemistry and biology focused on application of pharmacophore-directed retrosynthesis to assist with cellular mechanism of action studies of bioactive natural products, synthetic methodology focused on new organocascade processes including those directed toward the synthesis and application of β-lactones as intermediates for organic synthesis and exploring their utility as cellular probes and drug leads. |

|
Matthew Genzink completed a B.Sc. in Chemistry from Grove City College in 2018 where he performed research with Charles Kriley. During this time, he performed a research internship for three months at KU Leuven (Belgium) where he studied fluorescent probes with Wim Dehaen. In 2018 Matthew started at UW-Madison, joining the group of Tehshik Yoon where he was an NSF Graduate Research Fellow. Matthew currently studies Brønsted acid-catalyzed asymmetric photochemistry in the Yoon group. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved