Org. Synth. 2021, 98, 343-362
DOI: 10.15227/orgsyn.098.0343
Catalytic Diazoalkane-Carbonyl Homologation: Synthesis of 2,2-Diphenylcycloheptanone and Other Quaternary or Tertiary Arylalkanones and Spirocycles by Ring Expansion
Submitted by Jason S. Kingsbury,
1* Victor L. Rendina, Jacob S. Burman, and Brittany A. Smolarski
Checked by Junichi Taguchi, Haruka Fujino, and Masayuki Inoue
1. Procedure (Note 1)
2,2-Diphenylcycloheptanone (2). A three-necked 300-mL round-bottomed flask (central neck 29/32 joint, side necks 15/25 joint), equipped with a Teflon-coated magnetic stir bar (4.0 x 0.7 cm, rod-shaped) (Note 2) is charged with 1.0 g of scandium(III) triflate (Sc(OTf)3, 2.0 mmol, 5.6 mol%) (Note 3) directly in air. One side neck and the central neck of the reaction vessel are then fitted with glass stoppers. The other side neck is fitted with a short-path distillation head (15/25 upper outer joint, 15/25 lower inner joint, 15/25 side inner joint). While the distillation head is topped with a 15/25 three-way cock connected to an argon inlet and a vacuum line, its branch is fitted with a single-necked 25-mL round-bottomed flask (15/25 joint) charged with 2.00 g of phosphorous pentoxide (P2O5) (Note 4) to absorb the water of hydrated Sc(OTf)3. All joints are sealed with grease and Teflon tape (Figure 1A). After the entire apparatus is evacuated to 0.30 mmHg through the vacuum line, the flask is immersed in a silicone oil bath pre-heated to 150 °C. Keeping the reduced pressure below 0.30 mmHg, the catalyst powder is stirred at 150 °C for 12 h (Figures 1B and 1C). In the course of the drying, the formerly free-flowing white phosphorous pentoxide powder (Figure 1D) turns to a sticky white-pink solid containing phosphoric acid residues (Figure 1E).

Figure 1. (A) Set-up for drying commercial Sc(OTf)3 under a vacuum (150 °C, 0.30 mmHg); (B) Sc(OTf)3 before drying; (C) Sc(OTf)3 dried under a vacuum (150 °C, 0.30 mmHg) for 12 hours; (D) Phosphorous pentoxide before drying Sc(OTf)3; (E) Phosphorous pentoxide after drying Sc(OTf)3 (photos provided by checkers)
The assembly is next removed from the silicone oil bath, allowed to cool to 24 °C over the course of 20 min, and backfilled with argon. The distillation head is removed from the side-neck of the 300-mL flask, which is quickly fitted with a 15/25 three-way stopcock connected to an argon inlet and a vacuum line. Under argon flow, the two glass stoppers are removed from the flask. The uncapped side neck is fitted with a 15/25 rubber septum, and the central neck is fitted with a connecting adapter (upper outer joint 19/38, lower inner joint 29/32) and a 50-mL pressure-equalizing addition funnel, bearing an outer extra chamber for cooling (Note 5), which is topped with a 15/25 rubber septum. All junctures of the glassware are sealed with grease and Teflon tape. The flask is evacuated through the vacuum line (1.5 mmHg, 24 °C) for 1 min, after which the flask is backfilled with argon. This substitution of the gas is repeated three times. Then, the vacuum line is removed from the three-way cock, which is then connected to an argon outlet. Argon atmosphere is maintained throughout the reaction under a continuous argon flow. The assembly is positioned over a magnetic stir plate in a fume hood. (Figure 2A).

Figure 2. (A) Reaction set-up; (B) Reaction mixture after addition of toluene; (C) Reaction mixture after addition of cyclohexanone (photos provided by checkers)
To the flask is added toluene (32 mL to generate a 1.1 M solution) (Note 6) by a syringe through the rubber septum on the side neck. After suspending the catalyst in toluene (Figure 2B), 4.20 mL of cyclohexanone (3.98 g, 40.6 mmol, 1.13 equiv) (Note 7) is added by a syringe through the same rubber septum. The reaction mixture immediately turns from a white suspension to a pale yellow, homogeneous solution (Figure 2C). Then, the rubber septum on the top of the pressure-equalizing addition funnel is removed. In a separate single-necked 100-mL round-bottomed flask, 7.00 g of bright red diphenyldiazomethane (1, 36.0 mmol, 1.00 equiv) (Note 8) is dissolved in heptane (40 mL to generate a 0.90 M solution) (Note 9) at 0 °C, and the solution is transferred to the pressure-equalizing addition funnel using a 10-mL glass pipette. The rubber septum is reattached on the top of the funnel and sealed with Teflon tape. The solution inside the addition funnel, which is continuously cooled to 0 °C (Note 5) and shielded from light by aluminum foil, is added dropwise to the reaction mixture over 3 h at 24 °C (Notes 10 and 11) (Figure 3A). After the complete addition of the diazoalkane, the solution is allowed to stir for 3 h. The reaction mixture is light yellow in color but contains a brown, oily precipitate comprised of deactivated Sc(III) salts (Note 12) (Figure 3B).

Figure 3. (A) Diazoalkane addition to a solution of Sc(OTf)3 and substrate using a pressure-equalizing addition funnel; (B) The reaction mixture at the end of diazoalkane addition (photos provided by the checkers)
After the addition funnel and the three-way stopcock are removed, the heterogeneous mixture is diluted with diethyl ether (30 mL) (Note 13). The stir bar is removed, and the resulting reaction mixture is transferred to a 300-mL separation funnel. Additional diethyl ether (15 mL) is used to wash the flask. The resultant mixture is then washed with pure water (100 mL) (Note 14) (Figure 4A). The pale brown organic layer is separated, and the aqueous layer is extracted with diethyl ether (30 mL). The organic layers are combined into a 300-mL Erlenmeyer flask and dried over sodium sulfate (80 g) (Note 15) for 10 min (Figure 4B). The drying agent is filtered through a plastic funnel with a cotton plug into a 300-mL round-bottomed flask and rinsed with diethyl ether (2 x 5 mL). The filtrate is concentrated by rotary evaporation (38 °C, 35 mmHg) and dried on a high vacuum (room temperature, 1.5 mmHg) for 10 min. These workup operations furnish 10.8 g of pale brown translucent oil that contains the crude homologation product (Figure 4C).

Figure 4. (A) The organic layer and the aqueous layer; (B) The organic layer dried over Na2SO4; (C) The crude pale brown translucent oil (photos provide by the checkers)
Figure 5. (A) Purification by flash column chromatography; (B) Title compound 2 (photos provided by the checkers)
The crude oil is purified by flash column chromatography on silica gel using hexane and diethyl ether as eluents (Notes 16 and 17) (Figure 5A) to provide 5.34 g (56%) of desired product 2 as a pale-yellow solid (Note 18) (Figure 5B).
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the health hazards associated with
scandium(III) triflate,
phosphorous pentoxide,
toluene,
diphenyldiazomethane,
cyclohexanone,
heptane,
diethyl ether,
sodium sulfate,
hexane,
1,3,5-trimethoxybenzene, and silica gel.
2. All glassware and a Teflon-coated magnetic stir bar were oven-dried at 120 °C for 2 h and then cooled to room temperature in a desiccator cabinet prior to reaction set-up. The room temperature throughout this manuscript refers to temperature between 24 °C to 26 °C.
3.
Sc(OTf)3 (99%) was purchased from Sigma-Aldrich and dried rigorously using the apparatus pictured in Figure 1A. Higher grade lots of catalyst (99.995% trace metals basis) are available from multiple vendors, but the submitters found that all commercial supplies contained variable amounts of water due to the hygroscopic nature of
Sc(OTf)3. Attempts to use
Sc(OTf)3 in hydrated form (as received) can be met by substantial decomposition of the diazoalkanes, presumably due to the formation of triflic acid
in situ. Once dry, the
Sc(OTf)3 powder weighed only 0.88 g by mass difference (1.8 mmol, catalyst loading of 5.0 mol % relative to the limiting reagent).
4.
P2O5 (ACS Reagent-grade, >98%) was purchased from Sigma-Aldrich and used as received (submitters).
P2O5 (Extra Pure Reagent, >97.0%) was purchased from Nacalai Tesque, Inc. and used as received (checkers).
5. The outer chamber of the pressure-equalizing funnel is connected to a chiller, purchased from Tokyo Rikakikai Co., Ltd, via inlet and outlet Teflon tubes insulated with polyurethane foam. Methanol is used as a circulating refrigerant, and the cooling temperature is kept at 0 °C.
6. PRA-grade
toluene (99.8%) was purchased from Sigma-Aldrich and stored over vacuum-dried 3Å molecular sieves prior to use (submitters).
Toluene (purity GC 99.9%) was purchased from FUJIFILM Wako Pure Chemical Corporation and purified by Glass Contour solvent dispensing system (Nikko Hansen & Co., Ltd.) (checkers).
7.
Cyclohexanone was purchased from Sigma-Aldrich and stored over vacuum-dried 3Å molecular sieves prior to use (submitters).
Cyclohexanone (99.8%) was purchased from Sigma-Aldrich and used as received (checkers).
8.
Diphenyldiazomethane (
1) was prepared by applying Javed and Brewer's method described in
Org. Synth. 2008,
85, 189-195.
2b Purity of
1 was determined to be 97.1% by qNMR using 16.1 mg of
1 and 15.2 mg of
1,3,5-trimethoxybenzene (FUJIFILM Wako Pure Chemical Corporation, >99.0%, used as received) as an internal standard, with relaxation time (D
1) set to 30 seconds (checkers).
9. HPLC-grade
heptane was purchased from Pharmco-Aaper and used as received (submitters).
Heptane (Anhydrous, 99%) was purchased from Sigma-Aldrich and used as received (checkers).
10. The submitters reported that not only a pressure-equalizing addition funnel but also a Razel R-99 syringe pump with a setting of '1.5' on the adjustable dial was available for the purpose of slow addition of
1 (Figure 6). The solution of
1 in
heptane was pulled into a disposable gas tight syringe fitted with a 12-inch, 18 gauge needle and added to the reaction mixture at a rate of 10 mL/min at 23 °C.
Figure 6. Syringe pump diazoalkane addition to a solution of Sc(OTf)3 (photo provided by submitters)
11. The most important feature of the slow addition, which helps to offset bimolecular decomposition of diazoalkane
1, is that it be performed at a rate that is convenient but avoids any buildup of red color within the reaction mixture. The rate of productive diazo consumption (homologation vs. dimerization) may change as the reaction progresses.
12. TLC analysis of the reaction mixture is shown below. R
f values of benzophenone (side product) and diphenylcycloheptanone (desired product,
2) in
hexane/
diethyl ether (10/1, v/v) are 0.40 and 0.34, respectively. These spots can be viewed by fluorescence on silica gel 60 F
254 plates (TLC Silical gel 60 F
254, purchased from Merck KgaA) with UV light (254 nm) (Figure 7). The desired product
2 can be also stained with phosphomolybdic acid (PMA, FUJIFILM Wako Pure Chemical Corporation).
Figure 7. TLC analysis of the reaction mixture (photo provided by checkers)
13. Uninhibited (ethanol-free) ACS reagent-grade
diethyl ether was purchased from Pharmco-Aaper and used as received (submitters).
Diethyl ether (>99.0%) was purchased from FUJIFILM Wako Pure Chemical Corporation and used as received (checkers).
14. Pure water (≤100%) was purchased from FUJIFILM Wako Pure Chemical Corporation and used as received (checkers).
15. Anhydrous
Na2SO4 (>99.0%) was purchased from Nacalai Tesque, Inc. and used as received (checkers).
16. Flash column chromatography is performed on silica gel (Kanto Chemical Co., Inc., Silica gel 60 N, spherical and neutral, 0.040-0.050 mm) using compressed air. The mobile phase was purchased from Kanto Chemical Co., Inc. (
hexane) and FUJIFILM Wako Pure Chemical Corporation. (
diethyl ether) and used as received. The column with a 7.5 cm diameter x 15 cm height is wet packed with 300 g of silica gel in
hexane (500 mL). Sea sand is added to the bottom and to the top of the column. The crude oil is loaded onto the column using 15 mL of
toluene (>99.5%, FUJIFILM Wako Pure Chemical Corporation, used as received) (Figure 5A). At this point, fraction collection (250 mL x 20) is begun, and elution is continued with 5000 mL of
hexane/
diethyl ether (30/1, v/v). The desired product is obtained in fractions No. 14 through No. 17 as a pure form (Figure 8A). The fractions No. 9 through No. 13 are co-elution of the desired product
2 containing benzophenone. These impure fractions are combined and concentrated by rotary evaporation (38 °C, 15 mmHg). The obtained yellow oil is purified again using column chromatography. The column with a 5.0 cm diameter x 15 cm height is wet packed with 150 g of silica gel in
hexane (300 mL). Sea sand is added to the bottom and to the top of the column. The crude oil is loaded onto the column using 10 mL of
toluene. At this point, fraction collection (50 mL x 60) is begun, and elution is continued with 1750 mL of
hexane/
diethyl ether (30/1, v/v) and then 1250 mL of
hexane/
diethyl ether (15/1, v/v). The desired product is obtained in fractions 33-52 as a pure form (Figure 8B). These fractions and the fractions 14-17 of the first purification are combined, concentrated by rotary evaporation (38 °C, 15 mmHg), and dried on a high vacuum (room temperature, 1.5 mmHg) for 4 h.

Figure 8. TLC analysis of column chromatography (A) First purification; (B) Second purification (checkers). The spots on the silica gel plate are visualized under UV light (254 nm) (photos provided by checkers)
17. The desired product (
2,2-Diphenylcycloheptanone,
2) shows the following characteristics: mp = 92-95 °C; IR (cm
-1) 3057, 2932, 2858, 1704, 1598, 1495, 1444, 1319, 1156, 1034, 751, 700;
1H NMR
pdf (400 MHz, CDCl
3) δ: 1.72-1.73 (m, 4H), 1.78-1.80 (m, 2H), 2.59-2.66 (m, 4H), 7.15-7.30 (aromatic, 10H);
13C NMR
pdf (100 MHz, CDCl
3) δ: 24.9, 26.5, 30.6, 37.3, 42.4, 65.0, 126.5 (2C), 128.0 (4C), 128.7 (4C), 144.3 (2C), 212.5; HRMS Calcd for C
19H
20O Na, [M+Na]+: 287.1406, Found 287.1406; Anal. Calcd for C
19H
20O: C, 86.32; H, 7.63. Found: C, 86.14; H, 7.75; Purity was determined to be >98% by qNMR
pdf using 14.6 mg of
2 and 11.1 mg of
1,3,5-trimethoxybenzene (FUJIFILM Wako Pure Chemical Corporation, >99.0%) (checkers). Based on a purity of 98.5%, the amount of product formed is 5.25 g (55%).
18. A second run performed on the same scale provided
2 (5.30 g, 56% yield) in 99.0% purity as determined by qNMR.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. Diazo compounds are inherently toxic and irritating, especially on contact with the skin or eyes. Extreme caution and secondary containment must be applied when handling diazoalkane solutions, as there are constant risks associated with a spill or splash. Appropriate personal protective equipment (PPE), including safety glasses, a lab coat, and latex or nitrile gloves must be worn by practitioners. It is also prudent to manipulate diazo compounds behind a safety shield or with the sash of the fume hood lowered. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of according to local regulations. For general guidelines on the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The net or formal insertion of diazoalkyl carbon into non-polarized C-H and C-C bonds is a function of their proximity to carbonyl groups that engage in 1,2-addition/rearrangement. As such, the homologation of both aldehyde
3 and ketone
4 acceptors occurs with either a chain extension or ring expansion that is strategically powerful and often complexity-generating - especially in the case of substituted diazomethanes. For well over a century, the synthetic community has applied procedures for diazoalkane-carbonyl homologation that rely on traditional means for carbonyl activation, namely the presence of superstoichiometric amounts of B- and Al-based promoters
5 or general acid assistance based on water or alcohol co-solvents.
6 However, the past decade has seen a resurgence in the field of diazoalkane chemistry. The practicality of homologation reactions has improved with the development of Lewis acid catalysts that address previously unmet challenges, including low reactivity in the case of sterically hindered internal diazoalkanes, poor functional group tolerance, and greater regio- and stereochemical control.
In 2009, we reported a finding that commercial Sc(III) salts are uniquely potent catalysts for both ketone
7 and aldehyde
8 insertion reactions, focusing on application of non-carbonyl-stabilized diazo compounds as nucleophiles. A persistent goal in these investigations was to prepare both terminal and internal aryldiazomethanes without isolation using established methods for hydrazone oxidation.
9,10 As crystalline solids or high molecular weight oils, the stability of mono- and diaryldiazomethanes
11 is much higher than that of diazomethane - a poisonous, explosive gas that can only be handled under strict protocol as a dilute solution.
12 Reports show that diaryldiazomethanes have been applied industrially on-scale as donor-acceptor carbon sources for over twenty years without incident.
13 Procedure A was chosen to showcase the more challenging aspect of a diaryl quaternary carbon installation, yet it is applicable to many cycloalkanone ring sizes and a diversity of diazoalkane substituents (Table 1). α-Tertiary and -quaternary cycloalkanones
a-
j are all known substances that are fully characterized.
7 However, to our knowledge
ene-bicyclopentanones
3a and
3b are a,a-diphenyl ketones prepared by catalytic carbonyl homologation for the first time with Procedure A.
Table 1. Scope and Generality of Sc-Catalyzed Ring Expansion Reactions
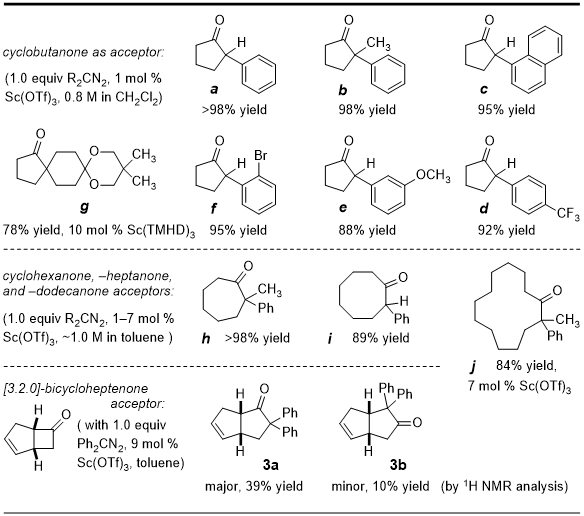
Additional discussion points on Sc-catalyzed ring enlargements include the following: (1) Different substitution patterns and electronic modifications (see
d-
f) in the aryldiazoalkane reaction partner are readily tolerated. Even
ortho-substituted nucleophiles give homologation products (
c,
f) in excellent yield; (2) The method also applies to aliphatic cyclic diazoalkanes and the production of spirocyclic structures. Synthesis of
g is an example revealing a tolerance for acid-labile ketal protecting groups and whose optimal 78% yield was achieved by switching to commercial Sc(TMHD)
3,
7a a more sterically hindered and less electron-poor catalyst; (3) Accommodation of further steric congestion is demonstrated by α-quaternary carbon installation with internal mono- and diarylated diazomethanes (→
b,
h,
j, and
2). The greater crowding inherent to disubstituted diazo compounds does not limit their reactivity to cyclobutanone, as unstrained electrophiles are also suitable; (4) Absent from Table 1 is an example of cyclopentanone homologation. Since the desired 2-arylcyclohexanones are more reactive than starting material, the reactions afford a mixture of products derived from over-homologation. Nonetheless, six-membered structures can be accessed from substituted lower homologs (
i.e.,
a-
f) by catalytic ring expansion with trimethylsilyldiazomethane. In such cases, 1,3-Brook rearrangement of α-trimethylsilyl cyclohexanone products gives enol silanes incapable of further reaction;
7b (5) Diphenyldiazomethyl insertion with commercial
cis-bicyclo[3.2.0]hept-2-en-6-one to give
3a and
3b in a 4:1 ratio illustrates a useful regioselectivity enabled by Sc(III)-catalysis. Predominant formation of
3a is consistent with a steric model14 in which the preferred 1,2-adduct (Sc-complexed diazonium betaine intermediate) situates the less substituted cyclobutane bond
anti-coplanar to the dinitrogen leaving group.
All arylcyclopentanone syntheses in Table 1 are complete in < 1 h at -78 °C with low catalyst loading (0.5-1 mol %) and are free of byproducts derived from dimerization of the diazo compound. Reactions involving the merger of mono-aryl and mixed alky/aryl-diazomethanes with higher cycloalkanones also take place rapidly at -78 to -48 °C but require higher catalyst loadings (7 mol %,
j, Table 1) for maximal efficiency. The optimal solvents for carbonyl homologation are
toluene and dichloromethane; acetonitrile can also serve to completely dissolve anhydrous
Sc(OTf)3 powder. Coordinating ethers such as tetrahydrofuran and
diethyl ether are less effective media, presumably due to competitive ligation to the trication in solution. Other strongly Lewis basic functions, such as free alcohols and amines, are not well tolerated, and O-H insertion products can be found for substrates containing hydroxyl groups. Some heteroaromatic functionality (
e.g. furans, thiophenes, but not pyridines) has proven compatible with the Sc-catalyzed homologation of aldehydes.
8bIn contrast to the features just mentioned, catalytic carbon insertion with diaryldiazomethanes is slower and best achieved at room temperature. The multigram synthesis of
2 (Procedure A) was optimized to permit subsequent testing of diazoalkane
415 as a carbon nucleophile for spirocyclic annulation. As illustrated below in Scheme 1,
spiro-fused dibenzo-bicycloundecanone
k is a known intermediate in a patented synthesis of a rigid
ene-ketone Michael acceptor showing μM affinity for mitochondrial permeability transition pore (MPTP).
16 As a polyprotein complex that regulates mitochondrial membrane permeability, MPTP has drawn attention as a mediator of energy metabolism, cellular Ca
2+ homeostasis, and programmed cell death (apoptosis). In the published route to
k, stepwise buildup of the quaternary α carbon and cyclo-pentanone ring was effected by a series of acidic and basic reaction conditions starting from the indicated dibenzonitrile (Scheme 1).
16 Therefore, we set out to test a direct, convergent synthesis of
k as a proving ground for Sc-catalyzed ring expansion despite the crowded and conformationally constrained nature of cyclic diaryldiazoalkane
4.
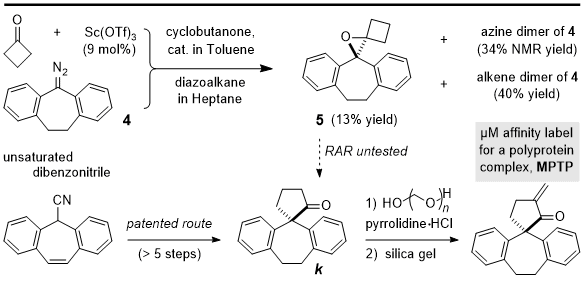
Scheme 1. Unusual Outcome in Targeting a Complex Spirocycle from 4
Unfortunately, slow addition of 5.6 g of the diazo compound (in heptane) to a premixed solution of 9 mol % catalyst and cyclobutanone (1 M in toluene) provided only the tetrasubstituted spirocyclic epoxide 5 in a low 13% yield following aqueous workup and chromatography. This outcome was reproducible over two separate runs involving the multigram scale, but in neither case was a cyclopentanone fraction detected by FTIR and 1H NMR analysis. Instead, the major products were the corresponding alkene (40%) and azine (34%) dimers. Herein lies a stark reminder that upper limits to the reaction's capability can persist even with the benefit of 4C ring strain in the electrophile. We believe that the energetically allowed mode of diazoalkyl addition to cyclobutanone orients the Sc(III)-alkoxide anti to the diazonium cation, with alignment favoring closure to the epoxy byproduct 5. Worse still, any bond rotation that would set up for the desired Wagner-Meerwein shift is associated with severe aryl/alkyl eclipsing interactions. Together with the fact that carbonyl 1,2-addition is reversible for highly resonance-stabilized diazo compounds, it is rather remarkable that strained spiroepoxide 5 could even be isolated. In our hands, the waxy material proved unpredictably labile depending on variables such as type of silica gel, time and temperature in solution, and exposure to trace impurities in solvents. It is possible that acidic conditions could be tested to effect rearrangement of 5 to the desired ketone k, but such efforts are arguably futile given the poor purified yield of 5.
Several nuances behind the known synthesis of
4 and its exploitation as a donor-acceptor carbon source speak further to its ultimate test on efficiency. For instance, its hydrazone precursor is indefinitely stable in the absence of moisture but is not accessible under standard conditions (H
2NNH
2, EtOH, reflux) from dibenzosuberone because of an unfavorable equilibrium. In an elegant solution by Miao and coworkers, the suberone is instead transformed first to the thioketone (with Lawesson's reagent) before condensation with hydrazine.
15 In this manner, hydrazone formation is driven to completion by release of gaseous H
2S. In the same report, diazoalkane
4 was applied in a two-step Barton-Kellogg synthesis of tetrabenzoheptafulvalene dimer; severe crowding in the fjord region around the central alkene led to discrete
syn and
anti stereoisomers at 23 °C.
15 Miao and coauthors advance such overcrowded bis-tricyclic ethylenes - the major product in our synthesis of
5 - as promising substructures for photo- and thermally-induceable molecular switches.
15 As a final example of anomalous behavior attributed to the dibenzocycloheptane framework, the azine dimer of
4 experiences complete hydrolytic breakdown on silica, leading to recovery of mostly dibenzosuberone during isolation of
5. Perhaps ongoing and continued tuning of lanthanide-based catalysts
17 for diazoalkane chemistry will be able to address the most elusive challenges of reactivity and regioselectivity in these transformations. In the meantime, we hope that our findings will only inspire and not deter other chemists from probing the upper limits of diazoalkane-carbonyl homologation, albeit with tempered expectations in structurally unusual or highly complex settings.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Scandium(III) trifluoromethansulfonate: Trifluoromethansulfonic acid, scandium(3+) salt; (144026-79-9)
Phosphorous pentaoxide: Phosphorous(V) oxide; (1314-56-3)
Cyclohexanone: Oxocyclohexane; (108-94-1)
Diphenyldiazomethane: Diazodiphenylmethane; (1) (883-40-9)
α,α-Diphenylcycloheptanone: 2,2-Diphenylcycloheptan-1-one (2)

|
Jason Kingsbury received his B.A. summa cum laude in Chemistry from Hamilton College and carried out undergraduate research with Professor Robin B. Kinnel, a veteran of marine natural products. From 1997-2003, he was an NSF predoctoral fellow in the labs of Amir H. Hoveyda at Boston College, where he synthesized new Ru-based catalysts for olefin metathesis. Following an NIH postdoctoral fellowship with E. J. Corey at Harvard University, he began his independent career in 2006 at Boston College before moving to California Lutheran University in 2013. His research interests include the design of new catalytic synthetic methods and strategies in targeting polycyclic natural products. |

|
Victor Rendina received a B.Sc. in Chemistry with distinction from The Ohio State University, where he performed undergraduate research under the direction of Professor T. V. RajanBabu. In 2008, he joined the Kingsbury lab at Boston College, where he designed and tested tris(oxazoline) complexes in Sc(III)-catalyzed enantioselective diazoalkane-carbonyl homologation reactions. Upon receiving his Ph.D. in 2013, he went on to conduct process research in the pharmaceutical industry. In 2015, he worked to scale up energetic materials for a multi-national defense contractor. He is currently a lead software engineer for a smart home automation company in the greater Boston area. |

|
Jacob Burman was raised in Chatsworth, CA and entered California Lutheran University in 2011 to pursue a B.Sc. degree in Chemistry. In his junior and senior years, Jacob played a vital role in the Kingsbury group's transition from the east coast, setting up new lab space and furthering scale-up studies on catalytic diazoalkyl carbon insertion. In 2015, he joined Professor Simon Blakey's team at Emory University to specialize in organometallic chemistry. His Ph.D. studies led to complementary methods of preparing regioisomeric allylic amines via Group IX MCp*-π-allyl intermediates. In 2019, he joined a discovery synthesis group at Raybow Pharmaceuticals in Brevard, North Carolina. |

|
Brittany Smolarski was born in Thousand Oaks, CA and entered California Lutheran University as a Biology major. Her focus switched to Chemistry after taking organic chemistry in her sophomore year with Professor Kingsbury. She performed over two years of research as a Swenson and John Stauffer fellow, designing a new entry to dialkyl-aminonaphthylpyridium (DANPY) fluorophores and helping to optimize the article's title reaction. She graduated with distinction in 2016 and then joined Professor Dale Boger's lab at The Scripps Research Institute (La Jolla, CA). She is now a law student at the University of San Diego. |

|
Junichi Taguchi was born in Saitama, Japan. He graduated from the University of Tokyo in 2021 with B.S. in Pharmaceutical Science. He is continuing his graduate studies at the University of Tokyo under the supervision of Prof. Masayuki Inoue. His research interests are in the area of the total synthesis of complex natural products. |

|
Haruka Fujino received his Ph.D. (2019) from The University of Tokyo under the supervision of Prof. Masayuki Inoue. During the Ph.D. course, he spent 2 months as Visiting Student at the University of Chicago under the direction of Prof. Scott A. Snyder (2016). After carrying out postdoctoral research with Prof. Seth B. Herzon at Yale University (2019-2020), he was appointed as Assistant Professor in the Graduate School of Pharmaceutical Sciences at the University of Tokyo in 2020. His research interests include the total synthesis of bioactive and architecturally complex natural products. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved