Org. Synth. 2022, 99, 39-52
DOI: 10.15227/orgsyn.099.0039
Transition-Metal-Free Synthesis of an Aryl Boronate Ester through Base-Mediated Boryl Substitution of an Aryl Halide with a Silylborane
Submitted by Ryosuke Shishido
1a and Hajime Ito
1a,b*Checked by Praveen Kumar Gajula, Gopal Sirasani and Chris Senanayake
1. Procedure (Note 1)
4,4,5,5-Tetramethyl-2-(2,4,6-triisopropylphenyl)-1,3,2-dioxaborolane (1). A 500 mL, three-necked round-bottomed flask (Note 2), equipped with a Teflon-coated, oval magnetic stirrer bar (45 mm × 20 mm), and rubber septa, is charged with potassium methoxide (KOMe) (1.73 g, 24.7 mmol, 1.2 equiv) (Note 3) in a glove box under argon (Note 4). After the sealed reaction vessel is removed from the glove box, it is connected to a vacuum-nitrogen manifold through an inlet adapter and two rubber septa (Figure 1). 1,2-Dimethoxyethane (DME; 205 mL) (Note 5) is added by syringe. (Dimethylphenylsilyl)boronic acid pinacol ester [PhMe2Si-B(pin)] (8.09 g, 30.9 mmol, 1.5 equiv) (Note 6) is dissolved in DME (5 mL) and added by syringe through the septum. The resulting reaction mixture is stirred (650 rpm) for 10 min at 30 °C, which is the temperature of an oil bath. The color of the solution changes from white to yellow after 10 min (Figures 1a and 1b). 2-Bromo-1,3,5-triisopropylbenzene (5.81 g, 20.5 mmol, 1.0 equiv) (Note 7) is then added dropwise by syringe through the septum over 15 min, during which time the solution becomes cloudy (Figure 1c).

Figure 1. Color change of the reaction mixture over the course of reaction; (A) Before heating; (B) After stirring at 30 °C for 10 min; (C) After addition of 2-bromo-1,3,5-triisopropylbenzene (photos provided by checkers)
After stirring (650 rpm) the reaction mixture for 1 h at 30 °C (Note 8), the solution is cooled to 0 °C (temperature of an ice water bath) followed by slow addition of tetrabutylammonium fluoride (TBAF; 1.0 M THF solution, 33.0 mL, 33.0 mmol, 1.6 equiv) (Note 9) over 20 min (Figure 2). The resultant solution is stirred for 2 h at the same temperature.
Figure 2. Reaction mixture after addition of TBAF solution (photo provided by checkers)
Water (150 mL) is then added, and the resulting mixture is transferred to a 1-L separatory funnel and diluted with diethyl ether (150 mL). After extraction and separation, the aqueous phase is extracted with diethyl ether (3 × 150 mL). The combined organic layer is washed once with saturated aqueous NaCl (50 mL), dried over anhydrous MgSO4 (7.5 g) for 30 min, filtered through a funnel fitted with a filter paper, and concentrated on a rotary evaporator under reduced pressure (50 mmHg, 35 °C). The obtained residue is purified by flash chromatography on silica gel (Notes 10 and 11) and recrystallization from acetonitrile (Notes 12 and 13) to afford 4.84 g (14.6 mmol, 70.5%) (Note 14) of 4,4,5,5-tetramethyl-2-(2,4,6-triisopropylphenyl)-1,3,2-dioxaborolane (1) as a white solid (Notes 15 and 16) (Figure 3).
Figure 3. 4,4,5,5-Tetramethyl-2-(2,4,6-triisopropylphenyl)-1,3,2-dioxa-borolane (1) (photo provided by submitter)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
2-bromo-1,3,5-triisopropylbenzene,
(dimethylphenylsilyl)boronic acid pinacol ester,
potassium methoxide,
1,2-dimethoxyethane,
tetrabutylammonium fluoride,
tetrahydrofuran,
diethyl ether,
magnesium sulfate,
sodium chloride, silica gel,
hexane and
acetonitrile.
2. All glassware was oven-dried at 120 °C for 30 min and cooled to room temperature under vacuum (0.5 mmHg).
3.
Potassium methoxide (95%) was purchased from Sigma-Aldrich and used as received.
4.
Potassium methoxide was stored and handled in a glove box, as it is highly moisture-sensitive.
5.
1,2-Dimethoxyethane (>99.0%) was purchased from Tokyo Chemical Industry (TCI) and distilled from sodium-benzophenone ketyl.
6.
(Dimethylphenylsilyl)boronic acid pinacol ester (>95.0%) was purchased from TCI and used as received.
7.
2-Bromo-1,3,5-triisopropylbenzene (>95.0%) was purchased from TCI and used as received.
8. The reaction progress was followed by TLC analysis,
hexane/
diethyl ether (9:1). R
f (product) = 0.56.
9.
Tetrabutylammonium fluoride solution (1.0 M in
THF) was purchased from Sigma-Aldrich and used as received. The addition of
tetrabutylammonium fluoride solution was needed to remove unreacted
(dimethylphenylsilyl)boronic acid pinacol ester and the methoxydimethylphenylsilane byproduct.
10. Silica gel (silica gel 60N for flash chromatography, spherical, neutral, particle size; 40-100 µm),
hexane (95.0%), and
diethyl ether (99.0%) was purchased from Kanto Chemical Co. The checkers purchased
hexane (99.0%) and
diethyl ether (99.0%) from Oakwood Chemical and used the solvents as received.
11. Column chromatography was performed as follows: The crude material was dissolved in
hexane (5 mL), and loaded onto a column (diameter = 4 cm, silica height = 21.8 cm) of 140 g silica gel wetted with
hexane. The flask containing the crude material was then washed with 3 mL of hexane three times and the washings were loaded onto the column. The column was first eluted with
hexane (500 mL), then eluted with
hexane/
Et2O = 99.8:0.2 (500 mL),
hexane/
Et2O = 99.6:0.4 (500 mL),
hexane/
Et2O = 99.4:0.6 (500 mL),
hexane/
Et2O = 99.2:0.8 (500 mL),
hexane/
Et2O = 99.0:1.0 (500 mL),
hexane/
Et2O = 98.8:1.2 (500 mL) and
hexane/
Et2O = 98.6:1.4 (500 mL) using compressed air. Fractions of 100 mL were collected, and the desired product (
1) was contained in fractions 30-39 (the spot of the product overlapped that of an impurity in fractions 30-33) (Figures 4 and 5). Fractions 30-39 were combined and concentrated on a rotary evaporator (50 mmHg, 35 °C). The desired product (
1) was further purified by recrystallization because the chromatographed product (
1) contained a small amount of impurities (
Note 13).
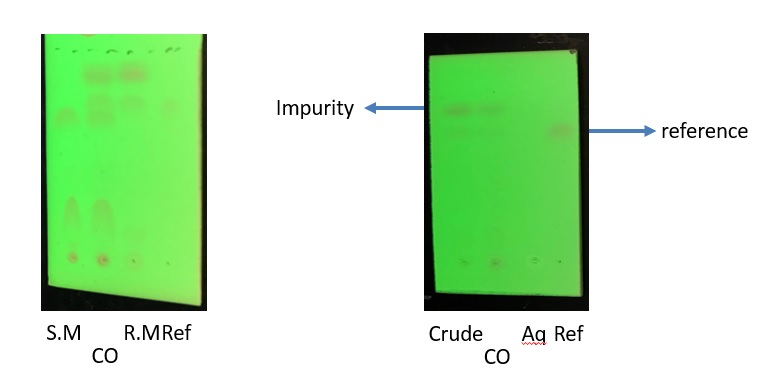
Figure 4. (A) Lanes )left-to-right) S.M (starting material); CO (co-spot of reaction mixture and product). R.M (reaction mixture-organic layer); Ref (Product reference). (B) Crude (reaction mixture crude-Organic Layer); CO (co-spot of reaction mixture and product); Aq (aqueous layer); Ref (Product reference) (photos provided by checkers)
Figure 5. TLC of the fractions after column purification (photo provided by checkers)
12.
Acetonitrile (>99.5%) was purchased from Kanto Chemical Co. and used as received. The checkers purchased
acetonitrile (>99.0%) from Oakwood Chemicals and used as received.
13. Recrystallization was performed as follows: Crude product
1 and
acetonitrile (15 mL) were added into a 100 mL, one-necked round-bottomed flask. The mixture was gently heated until all the solids dissolved (around 65 °C) (Figure 6). Then, the flask was stoppered and put into a freezer (-16 °C) to precipitate white solids. After 12 h the white solids were collected by vacuum filtration and washed with ice-cooled
acetonitrile (1.5 mL)
Figure 6. The mixture at (around 65 °C) all the solids were dissolved during recrystallization (photo provided by checkers)
14. A second reaction on half scale provided 2.32 g (7.02 mmol, 68.6%) of the same product with >98% wt. purity.
15. Characterization data for product
1;
1H NMR
pdf (400 MHz, CDCl
3) δ: 1.23 (d,
J = 6.9 Hz, 6H), 1.27 (d,
J = 7.0 Hz, 12H), 1.37 (s, 12H), 2.86 (septet,
J = 6.9 Hz, 1H), 2.98 (septet,
J = 6.9 Hz, 2H), 6.85 (s, 2H).
13C NMR
pdf (101 MHz, CDCl
3) δ: 24.1 (
CH
3), 24.6 (
CH
3), 25.1 (
CH
3), 34.1 (
CH), 34.6 (
CH), 83.7 (
C), 119.7 (
CH), 149.8 (
C), 152.0 (
C). The carbon directly attached to the boron atom was not detected, likely due to quadrupolar relaxation. 11B NMR
pdf (127 MHz, CDCl
3) δ: 32.8. Boron trifluoride diethyl ether was used as an external standard. FT-IR (ATR): 2955, 2867, 1605, 1556, 1459, 1382, 1354, 1329, 1291, 1214, 1143, 1101, 1065, 962, 857, 691 (submitter's data). HRMS-EI (
m/z): [M]
+ calcd for C
21H
3510B
16O
2, 329.27664; found 329.27625 (submitter's data).
16. The purity of the product (
1) was determined to be >98% wt. by quantitative
1H NMR
pdf spectroscopy in CDCl
3 using 9.23 mg of the product (
1) and 12.14 mg of 1,3,5-trimethoxybenzene as an internal standard.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Organoboron compounds are highly versatile building blocks in modern organic chemistry because they can be readily transformed into a wide variety of different functional groups.
2 Furthermore, they are easily handled because of their modulable reactivity and high air- and moisture-stability. Reactions of boron electrophiles with carbon nucleophiles such as Grignard reagents or organolithium reagents are popular methods for the preparation of organoboron compounds.
2 However, these methods suffer from low functional compatibility because of their strong basicity and nucleophilicity. In recent years, many transition-metal-catalyzed borylation reactions have been reported, exhibiting broad functional group compatibility.
3,4 However, using these methods to prepare pharmaceuticals is undesirable because of the high cost of the catalyst and the potential for contamination of the product with residual transition-metal impurities. Several transition-metal-free borylation methods have also been reported,
5,6,7,8 but these reactions still have issues in terms of their reactivity, regioselectivity and functional group compatibility.
In this context, we developed a formal nucleophilic boryl substitution reaction of organohalides with silylborane PhMe
2Si-B(pin) and an alkoxy base [
i.e., base-mediated borylation with silylborane (BBS method)].
9 This reaction proceeds smoothly without transition-metal catalysts to provide the borylated product in excellent yield with high borylation/silylation selectivity. Screening various conditions identified the optimum method was reaction of aryl halides with PhMe
2Si-B(pin) (1.5 equiv), and KOMe (1.2 equiv) in
1,2-dimethoxyethane (
DME) at 30 °C for one hour (Table 1). The use of the less bulky alkoxy base and ethereal solvent was important to ensure a good yield and borylation/silylation selectivity. This boryl substitution reaction was counterintuitive because a silylborane generally reacts with a base to generate a silyl nucleophile rather than a boryl nucleophile, and the generation of a boryl nucleophile from a silylborane has never been reported.
5c,10 This reaction exhibits high functional group compatibility, and can even be applied to substrates bearing ester and amide functional groups. Sterically demanding substrates also undergo the reactions effectively. Selected examples of substrates for this reaction of aryl-, heteroaryl- and alkyl halides are shown in Table 1.
Table 1. Representative borylated products synthesized via base-mediated borylation with silylborane (BBS method)a
Unfortunately, this reaction is not suitable for substrates with electrophilic or protic functional groups. Some incompatible substrates are shown in Table 2.
Table 2. Incompatible aryl bromide substratesa
Based on experimental observations and DFT studies using an artificial force-induced reaction (AFIR) method, we propose a carbanion-mediated mechanism for the BBS method (Scheme 1).
9 PhMe
2Si-B(pin) initially reacts with the alkoxide base KOMe to furnish the ate complex
A. Subsequent nucleophilic attack of the silyl moiety in complex
A on the bromine atom in the phenyl bromide leads to the formation of the carbanion species
B. Subsequently, the carbanion species attacks the boron electrophile rather than the silyl bromide to give the corresponding organoborate salt
C, which reacts with the silyl bromide to afford the phenylboronate ester.
Scheme 1. Proposed reaction mechanism of boryl substitution of Ph-Br with PhMe2Si-B(pin)/KOMe
Appendix
Chemical Abstracts Nomenclature (Registry Number)
2-Bromo-1,3,5-triisopropylbenzene: Benzene, 2-bromo-1,3,5-tris(1-methylethyl)-; (21524-34-5)
2-(Dimethylphenylsilyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane: 1,3,2-Dioxaborolane, 2-(dimethylphenylsilyl)-4,4,5,5-tetramethyl-; (185990-03-8)
Potassium methoxide: Methanol, potassium, salt (1:1); (865-33-8)
1,2-Dimethoxyethane: Ethane, 1,2-dimethoxy-; (110-71-4)

|
Ryosuke Shishido was born in Saitama, Japan, in 1991. He studied chemistry and received his B.S. degree (2015) and M.S. degree (2017) from Hokkaido University. Under guidance of Prof. Hajime Ito, he obtained his Ph.D. degree in 2020. His research interests include the synthesis of new silylborane reagents and the development of novel reactions with silylborane reagents. |

|
Hajime Ito was born in Osaka, Japan, in 1968. He completed his Ph.D. degree in 1996 under the direction of the late Professor Yoshihiko Ito. He then worked as an Assistant Professor at Tsukuba University in collaboration with Professor Akira Hosomi, and then moved to the Institute for Molecular Science. He then joined Professor Kim D. Janda's research group at the Scripps Research Institute as a research associate in 2001. In 2002 he was appointed as an Associate Professor at Hokkaido University, working with Professor Masaya Sawamura. He was promoted to a full Professor at the same university in 2010. |

|
Praveen Kumar Gajula received his Bachelor's and Master's degrees from Hyderabad. He obtained his Ph.D. in synthetic organic chemistry in 2012 from CSIR-IICT, Hyderabad under the guidance of Dr. Tushar Kanti Chakraborty. His research was focused on amide-modified RNA mimics, total syntheses of natural products, anti-cancer compounds and analogs thereof. In 2019, he joined Prof. Eriks Rozner's research group at Binghamton University, studying the optimization of RNA interference (RNAi) and CRISPR efficiency and specificity using chemically modified RNAs. In 2015, Praveen joined Sai Life Sciences, Hyderabad, India. He is currently a Senior Research Scientist at TCG GreenChem, Inc. in the department of process research and development. |

|
Dr. Gopal Sirasani received his Bachelor's and Master's degrees in Hyderabad, India. He obtained his Ph.D. in synthetic organic chemistry in 2011 from Temple University, Philadelphia under the guidance of Prof. Rodrigo B. Andrade. His doctoral research was focused on developing novel methodologies, total syntheses of natural products and analogs thereof. He got his post-doctoral training in the laboratory of Prof. Emily Balskus at Harvard University, where he developed biocompatible organic reactions utilizing microbially-generated reagents to realize transition metal catalysis in the presence of microbes. In 2013, Gopal began his industrial career at Melinta Therapeutics, New Haven, CT. He is currently working at TCG GreenChem, Inc. as a Director in the department of process research and development. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved