Org. Synth. 2022, 99, 53-67
DOI: 10.15227/orgsyn.099.0053
A Convenient Method for the Removal of Tetrabutylammonium Salts from Desilylation Reactions
Submitted by Luca McDermott, Dominick C. Witkowski, and Neil K. Garg*
1Checked by Philipp Spieß, Daniel Kaiser and Nuno Maulide
1. Procedure (Note 1)
4-Bromophenol (2). A single-necked (24/40 joint) 250 mL round-bottomed flask is equipped with a Teflon-coated magnetic stir bar (3.0 x 1.5 cm, football-shaped). The apparatus is flame-dried under reduced pressure and cooled to 23 °C under an atmosphere of nitrogen. The flask is then equipped with a rubber septum and then placed under positive pressure of nitrogen using a nitrogen inlet. To the flask is then added (4-bromophenol)(tert-butyl)dimethylsilane (1) (4.5 g, 3.8 mL, 16 mmol, 1.0 equiv) (Note 2) via syringe. Then, dry THF (30 mL) (Note 3) is added at 23 °C to the flask via syringe and stirring is started (300 rpm). After one min of stirring, tetrabutylammonium fluoride solution (19 mL, 1.0 M in THF, 19 mmol, 1.2 equiv) (Note 4) is added via syringe dropwise over five min (Figure 1A). The reaction mixture is then allowed to stir (300 rpm) at 23 °C for 30 min (Note 5). Then, the rubber septum is removed, and calcium carbonate (8.2 g, 82 mmol, 5.3 equiv) (Note 6) is added in one portion followed by Dowex 50WX8, 200-400 mesh, ion exchange resin (24 g) (Note 7) in two equal portions using a funnel to assist with the addition. To the reaction mixture is then added methanol (60 mL) (Figure 1B) (Note 8), the rubber septum is replaced, including the nitrogen inlet, and the suspension is stirred for 1 h at 23 °C (900 rpm).

Figure 1. A) Reaction setup after addition of TBAF; B) Reaction setup after addition of DOWEX ion-exchange resin
The resulting mixture is then filtered through a wetted celite pad (28 g, wetted with methanol (100 mL)) (Note 9) (7 x 4 cm) in a 150 mL medium porosity fritted Büchner funnel into a 1 L round-bottomed flask (24/40 joint) (Figure 2) using methanol (300 mL) as the eluent (Note 10).
Figure 2. Filtration apparatus
The filtrate is then concentrated by rotary evaporation (30 °C, 150 mmHg to 15 mmHg) under reduced pressure to yield a biphasic mixture of a colorless liquid and a yellow liquid.
The 1 L round-bottomed flask is then charged with silica gel (10.0 g) (Note 11). The crude material and silica gel are suspended in methylene chloride (60 mL) (Note 12) and concentrated under reduced pressure (30 °C, 375 mmHg to 22 mmHg) until a fine powder results. The product-adsorbed silica is then added to a column (7 cm OD x 20 cm tall) which is prepared using silica gel (165 g) that is wetted with pentane (500 mL) (Notes 13 and 14) (Figure 3A, 3B). The column is then eluted with 19:1 pentane:diethyl ether (2 L) (Note 15), followed by 9:1 pentane:diethyl ether (1 L), followed by 4:1 pentane:diethyl ether (1 L).
Figure 3. A) Product adsorbed silica gel; B) Column setup
The product is collected in 55 mL culture tubes, and the desired product elutes in fractions 35-66 (Note 16). These fractions are pooled and concentrated under reduced pressure (30 °C, 300 mmHg to 22 mmHg). The resulting yellow oil is then transferred to an 8-dram vial using diethyl ether and concentrated under reduced pressure (30 °C, 300 mmHg to 22 mmHg). Then, the resulting oil is dried under high vacuum at 23 °C for 18 h (Note 17) to yield a crystalline white solid (2.44 g, 88% yield, 98% purity) (Notes 18, 19, 20, and 21).
Figure 4. Isolated 4-bromophenol (2)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
(4-bromophenol)(tert-butyl)dimethylsilane,
tetrahydrofuran,
tetrabutylammonium fluoride solution,
calcium carbonate,
Dowex 50WX8, 200-400 mesh, ion exchange resin,
methanol, silica gel,
methylene chloride,
pentane,
diethyl ether, hexanes,
EtOAc, Celite,
CDCl3, and
1,3,5 trimethoxybenzene.
2.
4-Bromophenol)(tert-butyl)dimethylsilane (> 97%) was purchased from Sigma-Aldrich corp. and used as received.
3.
Tetrahydrofuran (1.0 M in
THF) (> 99.9%) was purchased from Fisher Scientific and used as received. The submitters passed it through an activated alumina column before use.
4.
Tetrabutylammonium fluoride (1 M solution in
THF) was purchased from Sigma-Aldrich corp. and used as received.
5. The progress of the reaction is monitored via TLC analysis on silica gel with 9:1 hexanes:
EtOAc used as the eluent. Hexanes (>98.5%) and
EtOAc (98.5%) were purchased from Fisher Scientific and used as received. The plate is visualized using a UV lamp (254 nm).
Figure 5. TLC of the crude reaction mixture after 30 minutes. SM = starting material (starting material Rf: 0.8), CS = co-spot of starting material and reaction mixture, RXN = reaction mixture (product Rf: 0.2)
6.
Calcium carbonate (> 99%) was purchased from Fisher Scientific and used as received.
7.
DOWEX 50WX8 200-400 mesh ion exchange resin was purchased from Sigma-Aldrich corp. and used as received.
8.
Methanol (> 99.8%) was purchased from Fisher Scientific and used as received.
9. Celite was purchased from Fisher Scientific and used as received.
10. The reaction flask was rinsed three times with
methanol (100 mL) to achieve a quantitative transfer of the contents of the reaction flask through the celite pad.
11. SiliaFlask P60 (particle size 0.040-0.063 nm) was purchased from SiliCycle and used as received.
12.
Methylene chloride (> 99.5%) was purchased from Fisher Scientific and used as received.
13.
Pentane (98%) was purchased from Honeywell and used as received. The submitters used hexanes instead of
pentane.
14. To assist with quantitative transfer of product-adsorbed silica onto column, sand is added to the round-bottomed flask and swirled to remove the silica from the side of the flask. This sand is then added to the column after the product-adsorbed silica.
15.
Diethyl ether (> 99%) was purchased from Fisher Scientific and used as received.
16. Fractions containing the product were identified by TLC analysis using 9:1 hexanes:
EtOAc as the eluent where
2 has an R
f of 0.20. Fractions 35-66 contained the desired product, and each fraction is rinsed with Et
2O (2 x 1 mL) to ensure quantitative transfer.
Figure 6. TLC analysis of column fractions
17. Prior to drying at 23 °C, the submitters submerged the dram vial containing the oil in a dry ice-acetone bath (-78 °C) and dried under high vacuum (<1 mmHg) for 1 h. The checkers omitted this step and obtained the same result.
18. To assist with crystallization, crystals on the side of the vial can be placed in the oil, either by tilting the vial such that the crystals fall into the oil, or by scraping them into the oil with a spatula. The submitters observed significantly delayed crystallization that was assisted by brief additional cooling (-196 °C liquid
nitrogen for ~30 sec) after drying.
19. The product can be characterized as follows:
1H NMR
pdf (400 MHz,
CDCl3) δ: 7.32 (d,
J = 8.6 Hz, 2H), 6.71 (d,
J = 8.6 Hz, 2H), 5.21 (s, 1H);
13C NMR
pdf (101 MHz,
CDCl3) δ: 154.48, 132.63, 117.33, 113.16.; IR (film): 3334, 1487, 1217, 824 cm
-1; HRMS-ESI (
m/z) [M-H] calcd for C
6H
4OBr, 170.9451; found, 170.9451; mp 61-63 °C; R
f 0.20 (9:1 hexanes:
EtOAc).
20. A second run on half scale was performed, affording 1.29 g of
2 (94% yield, 98% purity). The submitters obtained 2.52 g (93% yield, >97% purity) and 2.37 g (87% yield, >97% purity) of
2 in two separate full-scale runs.
21. The purity of
2 was determined by qNMR
pdf in all cases, using
1,3,5 trimethoxybenzene (Sigma-Aldrich, >99.9%) as the external standard.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Silicon-based protecting groups are commonplace in synthetic routes to complex organic molecules.
2 Their ability to be introduced under mild conditions, as well as their stability toward harsh reaction conditions, has made them a widespread tool in organic synthesis. Small silyl protecting groups, such as the trimethylsilyl group, can be removed under basic or acidic conditions, but larger silyl protecting groups are generally removed under fluoride-mediated conditions. One especially useful reagent for the removal of silyl protecting groups is
tetrabutylammonium fluoride (
TBAF).
3 The solubility of this reagent in organic solvents, as well as its chemoselectivity, has contributed to its frequent usage in the removal of silyl protecting groups. On simple systems, where the products are relatively non-polar,
TBAF is easily removed via an aqueous workup. For more polar products, however, an aqueous workup can result in loss of material to the aqueous layer or challenging separations.
An attractive alternative protocol for the removal of residual
TBAF was reported by Kishi and co-workers in 2007.
4 In this study, removal of these byproducts was achieved without an aqueous extraction, and applied in the total synthesis of Halichondrin B. In particular, it was discovered that the addition of a 50WX8 ion-exchange resin in combination with a mild base, such as
calcium carbonate, was effective in sequestering cationic byproducts, such as
tetrabutylammonium, from reaction mixtures. The proposed mechanism for this methodology is delineated in Figure 7. First, the
tetrabutylammonium cation exchanges with a proton on the ion-exchange resin. This equilibrium process is then driven forward by
calcium carbonate, which reacts with HF to form calcium fluoride, water, and carbon dioxide. After addition of the ion-exchange resin and
calcium carbonate, the reaction mixture can be filtered to provide the desired product. This convenient and effective procedure removes undesired
TBAF-related byproducts and obviates the need for aqueous extraction.
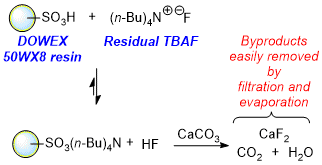
Figure 7. Mechanism of TBAF removal by DOWEX 50WX8 resin
The Kishi group explored the utility of this methodology in the syntheses of several alcohol products (Figure 8). It was observed that carbohydrates, such as 3, could be isolated from their respective tert-butyl silyl (TBS)-protected precursors. Furthermore, diols such as 4 and 5 could also be isolated from the corresponding silyl alcohols using this methodology.
Figure 8. Examples of desilylated alcohol products accessed using the DOWEX 50WX8 resin method
This strategy was also employed in the total synthesis of complex, highly polar natural products (Figure 9). For example, penta-ol
6 was accessed in near quantitative yield and served as a key late-stage intermediate in Kishi's total synthesis of halichondrin B.
4 The natural product alloviroidin (
7) was also isolated following the removal of five TBS groups in the final step of the total synthesis,
5 and tri-ol intermediate
8 was accessed using this methodology en route to etnangien.
6 Finally, tetra-ol
9 was isolated and further derivitized en route to zincophorin methyl ester.
7 Our group has also found this methodology to be useful for removing
TBAF related byproducts from polar intermediates during total synthesis campaigns. Overall, the ion-exchange resin strategy for the removal of
TBAF related byproducts has shown to be a convenient and effective workaround to challenges encountered in the desilylation of alcohols and purification of the resultant polar products.
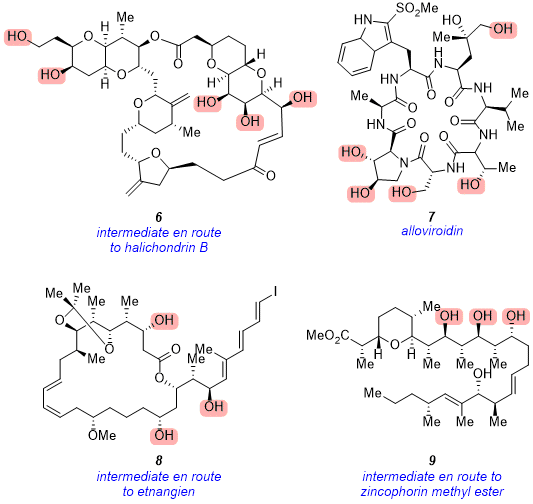
Figure 9. Complex late-stage alcohol intermediates and natural products accessed using the DOWEX 50WX8 resin method. Alcohols highlighted in pink were revealed using this method.
Herein, we demonstrate the use of this resin to remove TBAF following the multi-gram scale deprotection of a TBS-protected phenol. In this procedure, no aqueous workup was required, and 1H NMR analysis of the crude material showed complete removal of TBAF. Indeed, the only impurities that remain after ion-exchange resin workup are derived from the silyl protecting groups. In some cases, such byproducts can be removed simply by evaporation under vacuum. However, on this scale, we observed that column chromatography followed by vacuum evaporation is required to remove all of the silyl byproducts. Overall reaction time, including both desilylation and treatment with DOWEX resin, was less than 2 hours, and near quantitative yield of the desired product was observed. This simple procedure provides an attractive solution for the purification of highly polar molecules following removal of silyl protecting groups with TBAF.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
4-Bromophenol)(tert-butyl)dimethylsilane; (1) (67963-68-2)
Tetrabutylammonium fluoride solution 1.0 M in THF; (429-41-4)
Calcium carbonate; (471-34-1)
DOWEX 50WX8, 200-400 mesh, ion exchange resin; (69011-20-7)

|
Luca McDermott was born and raised in San Francisco, CA. In 2020, he received his B.S. in Biochemistry from Tufts University where he carried out research under the direction of Professor Clay S. Bennett. In 2020 he began graduate studies at the University of California, Los Angeles, where he is currently a second-year graduate student in Professor Neil K. Garg's laboratory. His dissertation studies are primarily focused on total synthesis. |

|
Dominick Witkowski was born in Portsmouth, VA and raised in Northborough, MA. In 2020, he received his B.A. in Chemistry from Boston University where he carried out research under the direction of Professor John A. Porco, Jr. He then moved to the University of California, Los Angeles where he is currently a second-year graduate student in Professor Neil K. Garg's laboratory. His studies primarily focus on developing synthetic methods utilizing strained cyclic allenes. |

|
Neil Garg is a Distinguished Professor of Chemistry and the Kenneth N. Trueblood Endowed Chair at the University of California, Los Angeles. His laboratory develops novel synthetic strategies and methodologies to enable the total synthesis of complex bioactive molecules. |

|
Philipp Spieß conducted his B.Sc. and M.Sc. studies at the University of Munich (LMU Munich) and finished his M. Sc. degree with a research stay in the group of Prof. Ruben Martin (ICIQ, Tarragona), working on nickel catalysis. In 2020, he moved to the University of Vienna to undertake Ph. D studies in Prof. Nuno Maulide's laboratory. His studies primarily focus on total synthesis and on developing new methodologies in the area of amide activation. |

|
Daniel Kaiser received his Ph.D. at the University of Vienna in 2018, completing his studies under the supervision of Prof. Nuno Maulide. After a postdoctoral stay with Prof. Varinder K. Aggarwal at the University of Bristol, he returned to Vienna in 2020 to assume a position as senior scientist in the Maulide group. His current research focusses on the chemistry of destabilized carbocations and related high-energy intermediates. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved