Org. Synth. 2022, 99, 92-112
DOI: 10.15227/orgsyn.099.0092
Preparation of MIDA anhydride and Reaction with Boronic Acids
Submitted by Peng-Jui Chen, Aidan M. Kelly, Daniel J. Blair, and Martin D. Burke*
1Checked by Jack Hayward Cooke and Richmond Sarpong
1. Procedure (Note 1)
A. MIDA anhydride (1). A 500 mL single-necked, 24/40 round-bottomed flask equipped with a 5 x 2 cm Teflon-coated magnetic stirring bar is charged with methyliminodiacetic acid (40.0 g, 270 mmol, 1.00 equiv)(Notes 2 and 3), capped with a rubber septum and evacuated and backfilled with nitrogen via 20 G needle. Acetic anhydride (140 mL, 1.49 mol, 5.52 equiv) (Note 4) is added via syringe as a single portion to form a colorless suspension. This is immediately followed by the addition of pyridine (3.30 mL, 40.5 mmol, 0.15 eq.)(Note 5) in a single portion (Figure 1B). The flask is stirred under nitrogen in an oil bath at 70 °C for 1.5 h (Figure 1C), at which time analysis of an aliquot of the reaction mixture by 1H NMR in DMSO-d6 indicates complete consumption of MIDA.

Figure 1. A) Drying MIDA using reduced pressure prior to reaction (see Note 2); B) Reaction mixture after addition of pyridine and acetic anhydride; C) Heating reaction mixture at 70 °C using an oil bath (photos provided by submitters)
A brown homogeneous solution forms after 1.5 h (Figure 2A). Upon cooling to room temperature, the mixture is carefully concentrated (to avoid bumping the insoluble material) by direct rotary evaporation (37 °C/2.4 mmHg) of the reaction flask. The remaining acetic anhydride, acetic acid, and pyridine are removed through a toluene azeotrope (12 x 100 mL)(Note 6) using rotary evaporation (35 °C/2.4 mmHg). The brown residue (Figure 2B) is transferred portion wise to a 24/40 single-necked 1 L round-bottomed flask using multiple portions of diethyl ether (1 x 300 mL, 1 x 100 mL, and 1 x 100 mL)(Note 7). A 5 x 2 cm Teflon-coated magnetic stirring bar is added to the flask followed by activated carbon (10 g)(Note 8) as a single portion, and the solution stirred at room temperature for 15 min. The reaction mixture is filtered through a celite pad (2 cm) (Note 9) covered with sand (1 cm) using a coarse 8 cm glass frit into a 1 L Büchner flask (Figure 2C). The filter cake is washed with a single portion of diethyl ether (100 mL).

Figure 2. A) Reaction mixture after heating at 70 °C for 1.5 h; B) Crude mixture following azeotropic removal of volatiles; C) Appearance of crude material after treatment with activated carbon, filtration, and concentration (photos provided by submitters)
The colorless filtrate is transferred portion wise (3 x 200 mL portions) to a 500 mL single-necked 24/40 round-bottomed flask and concentrated by rotary evaporation (20 °C/200-300 mmHg) to afford a white solid (Figure 2D).
A reflux condenser is attached to the 24/40 single-necked 500 mL flask, which is immersed in an oil bath equilibrated to 40 °C, diethyl ether (30 mL) is added dropwise via syringe with stirring over 5 min, and left to stir for an additional 10 min at 40 °C. The flask is cooled to room temperature and left to stand at room temperature for 2 h. The condenser is removed and replaced with a 24/40 rubber septum, and the flask is then immersed in an ice bath for 30 min. The resulting solid is collected by filtration through a 4 cm coarse glass frit using a 250 mL Büchner flask to provide a white crystalline solid (23.6 g, 183 mmol, 68%) (Note 10) (Figure 3).
Figure 3. Isolated MIDA anhydride 1 following recrystallization from Et2O (photo provided by submitters)
B. 5-Acetylthiophene-2-boronic acid MIDA ester (2). A 500 mL single-necked, 24/40 round-bottomed flask equipped with a 5x2-cm Teflon-coated magnetic stirring bar is charged with 1 (20.1 g, 156 mmol, 3.00 equiv) (Note 11) and 5-acetylthiophene-2-boronic acid (8.84 g, 52.0 mmol, 1.00 equiv) (Note 12), capped with a 24/40 rubber septum, evacuated, and backfilled with nitrogen via 20 G needle. Dioxane (150 mL) (Note 13) is added via syringe as a single portion to form a brown suspension (Figure 4A). The round bottom flask is then heated in an oil bath at 70 °C for 24 h leading formation of a white precipitate (Figure 4B). Reaction completion is checked by TLC (Figure 4C). The reaction mixture is allowed to cool to temperature over 1 h, acetone (100 mL) is added, and the mixture is transferred to a 1 L separatory funnel. A single portion of ethyl acetate (100 mL) is used to rinse the reaction flask into the separatory funnel. Water (150 mL) is added, and the separatory funnel is shaken, then brine (50 mL) is added and the separatory funnel is gently inverted to assist phase separation. The organic layer is collected in a 1 L Erlenmeyer flask and the aqueous layer is extracted with ethyl acetate/acetone (9:1) (2 x 150 mL), using each wash volume to rinse the reaction flask into the separatory funnel between extractions (Note 14). The combined organic layers are dried over MgSO4 (100 g) for 15 min. The resulting solution is filtered in two equal portions through a solid addition funnel plugged with cotton wool into a 1 L single-necked 24/40 round-bottomed flask, and the solution is concentrated to dryness by rotary evaporation (25 °C/2.4 mmHg) to afford a brown paste (Figure 5A). The paste is further dried azetropically with toluene (2 x 50 mL) by rotary evaporation. Acetone (50 mL) (Note 15) is added as a single portion to the crude solid to create a fine suspension, and a spatula is used to loosen the solid from the sides of the round-bottomed flask. Diethyl ether (800 mL) is added to the 1 L round-bottomed flask with swirling, causing additional precipitation of the product, which is left to stand for 15 min. Solids are again freed from the sides of the flask with a spatula and the resultant pale brown precipitate is collected via vacuum filtration through a 8 cm coarse glass frit using a 2 L Büchner flask. The round-bottomed flask is washed into the filter funnel with additional protions of diethyl ether (2 x 100 mL) to provide the product as a pale brown solid (12.0 g, 42.7 mmol, 82%)(Note 16) (Figure 5B).

Figure 4. A) Reaction mixture prior to heating; B) Reaction mixture following heating at 70 °C for 24 h; C) TLC at 24 h (EtOAc) showing consumption of boronic acid (left channel) against the reaction mixture (right channel) with a co-spot (center channel) photos provided by submitters)
Figure 5. A) Crude reaction mixture after aqueous work-up and rotary evaporation; B) Isolated 5-Acetylthiophene-2-boronic acid MIDA ester (2) following precipitation from Acetone/Et2O (photos provided by submitters)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available
via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
methylamino diacetic acid,
acetic anhydride,
pyridine,
diethyl ether,
activated carbon,
5-acetylthiophene-2-boronic acid,
1,4-dioxane,
ethyl acetate, and
acetone.
2.
Methyliminodiacetic acid was purchased from Combi-Blocks (PN OR-0737) and was dried using a vacuum line (3.5 mmHg) for 16 h prior to use.
3.
Methyliminodiacetic acid can also be prepared according to Burke and co-workers,
Org. Synth. 2009,
86, 344, and must be dried using a vacuum line (3.5 mmHg) for 16 h prior to use.
4.
Acetic anhydride (99.5%) was purchased from Fisher Chemical (PN A10-500) and was used without any further purification.
5.
Pyridine (99%) was purchased from Alfa Aesar (PN 19378) was dried over CaH
2 and distilled at ambient pressure under N
2 prior to use.
6.
Toluene (99.9%) was purchased from Fisher Chemical (PN T324-4) and was used without any further purification.
7.
Diethyl ether (99%) was purchased from Fisher Chemical (PN E138-20) and was used without any further purification.
8.
Activated carbon was purchased from Fisher Scientific (PN D127-500) and was used without any further purification.
9. Celite was purchased from Fisher Chemical (PN C212-50LB) and was used without any further purification.
10. Repetition of this procedure on the same scale provided 23.3 g (67%) of
1. mp 42-44 °C;
1H NMR
pdf (500 MHz, DMSO
-d6) d: 2.31 (s, 3H), 3.60 (s, 4H)
; 13C NMR
pdf (126 MHz, DMSO
-d6) d: 42.4, 54.2, 165.8; IR (ATR Plate): 545, 589, 614, 946, 1108, 1209, 1251, 1265, 1337, 1457, 1763, 1779, 1812, 2798, 2954 cm
-1; HRMS (EI
+) for C
5H
7NO
3 [M+H]
+ calcd: 129.0426, found: 129.0426; LRMS (EI
+)
m/z (rel. intensity) 129.0 (M
+, 34%), 101.0 (5%), 57.1 (100%); Anal. Calc. C 46.51, H 5.46, N 10.85; found C 46.51, H 5.55, N 10.82.
11. Lower yields of
2 were observed by the submitters when using less than 3 equivalents of
1, 1.5 eq. (61%) and 2 eq. (59%).
12.
5-Acetylthiophene-2-boronic acid (98%) was purchased from Combi-Blocks (PN BB-2011) and was used without any further purification.
13.
Dioxane (99%) was purchased from Fisher Chemical (PN D111-500) and dried over activated 3 A molecular sieves (purchased from Alfa Aesar, PN L05335) and degassed using bubbling N
2 through a 20 G needle for 1 h prior to use.
14.
Ethyl acetate (99.5%) was purchased from Fisher Chemical (PN E145-20) and was used without any further purification.
15.
Acetone (99.5%) was purchased from Fisher Chemical (PN A18-20) and was used without any further purification.
16. Repetition of this procedure on half scale gave 5.8 g (79%) of
2. mp 225-227 °C;
1H NMR
pdf (500 MHz, DMSO
-d6) δ: 2.53 (s, 3H), 2.64 (s, 3H) 4.17 (d,
J = 17.2 Hz, 2H), 4.39 (d,
J = 17.2 Hz, 2H), 7.33 (d,
J = 3.7 Hz, 1H), 7.93 (d,
J = 3.7 Hz, 1H).;
13C NMR
pdf (126 MHz, DMSO
-d6) δ: 26.9, 47.6, 61.7, 134.1, 134.5, 147.1, 168.4, 190.6; IR (ATR Plate): 478, 596, 825, 897, 984, 1028, 1162, 1271, 1520, 1659, 1775, 2914, 2995 cm
-1;
11B NMR
pdf (128 MHz, DMSO
-d6) δ: 10.43; HRMS (ESI
+) for C
11H
13BNO
5S [M+H]
+ calcd: 282.0607, found: 282.0600; LRMS (ESI
+)
m/z (rel. intensity) 284.2 (2%), 283.2 (10%), 282.2 (M+H, 100%), 281.4 (18%), 148.2 (64%); Anal. Calc. C 47.0, H 4.29, N 4.98, S 11.41; found C 47.11, H 4.42, N 4.98, S 11.28.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Boronic acids are amongst the most widely used reagents for the assembly of small organic molecules.
2 However, their proclivity toward decomposition and idiosyncratic stability has limited their application in many complex settings.
3 A contributor to the unstable behavior of boronic acids is their Lewis acidic character, and in this regard, we identified that chelation of boron with
N-methyliminodiactic acid (
MIDA) coordinatively saturates and deactivates boronic acids to produce bench stable crystalline MIDA boronates, many of which are commercially available (Figure 6).
4Figure 6. Coordinative saturation of boronic acids through formation of MIDA boronates produces bench stable building blocks
Scheme 1. MIDA boronates robustly protect boronic acids against many common chemical transformations thereby facilitating the direct synthesis of complex boronic acid building blocks.
The protection of boron with
MIDA has broadly enabled the synthesis of many complex MIDA boronate building blocks using transformations otherwise incompatible with boronic acids (Scheme 1).
5 Notably, unlike many boronic acids MIDA boronates are stable toward column chromatography enabling these boronic acid precursors to be obtained in high purity.
The attenuation of boron reactivity afforded by
MIDA chelation is reversible, as MIDA boronates are easily cleaved
via the action of mild aqueous base to furnish the parent boronic acid.
6 Using this reversible attenuation of boron reactivity a strategy to perform iterative cross coupling analogous to iterative peptide synthesis has been developed, using halo-boronic acid building blocks (Figure 7). Here, instead of forming amide bonds, C-C bonds are formed in a sequential manner through iterative Suzuki-Miyaura cross-coupling reactions.
Figure 7. Iterative peptide coupling allows the synthesis of polypeptides through reversibly attenuating the reactivity of amines during amide bond formation by using fmoc or boc protecting groups. In an analogous process, MIDA reversibly attenuates the reactivity of boronic acids in Suzuki-Miyaura cross coupling reactions enabling iterative C-C bond formation
Ourselves and now many others have been able to utilize this iterative cross coupling strategy in the synthesis of natural products,
7 materials
8 and potential drug candidates.
9 The flexibility and modularity of this approach has facilitated the exploration of the biological function of complex natural products through the atomistic modification of the antifungal amphotericin B
10 as well as site selective
13C labelling of the anti-lipoperoxidant peridinin (Figure 8).
11Figure 8. Iterative assembly of MIDA boronate building blocks enables atomistic modification of complex natural product scaffolds to explore their structure-function relationships
Boronic acids possess vastly different stabilities which can be problematic when exposing these potentially sensitive compounds to the harsh conditions required to effect cross-coupling (Scheme 2). Combining
MIDA deprotection and coupling into one step it is possible to control the rate of release of a boronic acid over the course of a cross-coupling reaction such that competitive decomposition does not occur.
12 Other recent examples have enabled addition into sulfinyl aldimines,
13 speciation controlled multi-component cross coupling,
14 and 2-pyridyl cross couplings.
15Scheme 2. MIDA boronates increase the shelf life and cross-coupling efficiency of boronic acids
MIDA boronates possess an unusual binary affinity for silica gel (Figure 9). Specifically, MIDA boronates are minimally mobile on silica gel in Et
2O and rapidly eluted using THF agnostic of the appended alkyl fragment.
16 As a MIDA boronate is conserved during each cycle of iterative cross-coupling (Figure 3), this binary affinity for silica gel allows generalized small molecule purification similar to the catch-and-release strategies adopted in solid phase synthesis (Figure 6). During the "catch" step a crude mixture from an iterative cross-coupling reaction is loaded onto a silica gel cartridge. Washing with Et
2O/MeOH selectively elutes impurities without mobilizing the MIDA boronate product. Switching to THF mobilizes the MIDA boronate and elutes a solution of MIDA boronate in THF for further iterative cross-coupling.
Figure 9. MIDA boronates serve as a common functional handle for generalized purification via catch and release chromatography
Collectively these advances led to the creation of an automated synthesis platform where deprotection, cross-coupling and purification are operated by a self-contained synthesis machine.
16 In a demonstration of this technology the natural product ratanhine and 20 derivatives were assembled from a pool of 10 building blocks, representative examples are depicted in Figure 10. In each case the target molecule was obtained without modification of the reaction conditions or protocols.
Figure 10. Automated building block-based synthesis enabled the synthesis of 20 ratanhine derivatives from a suite of 10 building blocks without modification of coupling conditions. D = deprotection, C = cross-coupling, P = purification
In this report in Organic Syntheses we describe the synthesis of MIDA boronate building blocks using
MIDA anhydride 1,
17 a reagent which expands the range and simplifies the process of converting boronic acids into MIDA boronates. The most common method for generating MIDA boronates is based on reports from Contreras and co-workers
18 involving high temperature dehydrative condensation of the diacid
MIDA with a boronic acid (Scheme 3). Whilst these conditions have been effective in the preparation of a range of simple MIDA boronate building blocks, the high temperature acidic conditions are not compatible with sensitive boronic acids and have required the development of more specialized techniques,
19 particularly for the synthesis of heterocyclic substrates. Other methods for the synthesis of MIDA boronates boronic acids involve high temperature condensation using the free diacid or substrate specific conditions utilizing dihaloboranes.
20
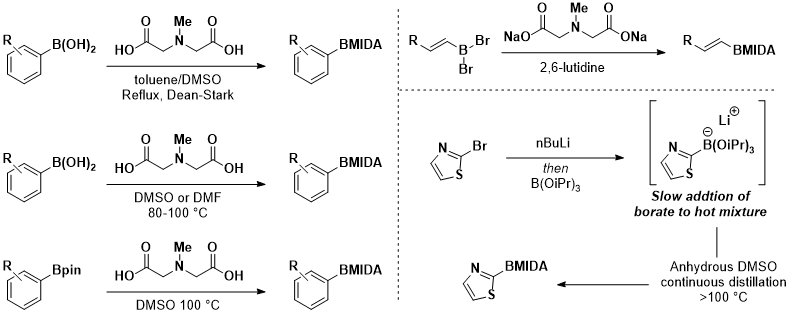
Scheme 3. Typical methods for the preparation of MIDA boronates
In contrast to these methods, heating a boronic acid with MIDA anhydride in anhydrous dioxane enables direct conversion of a boronic acid into a MIDA boronate, avoiding the high temperature acidic conditions associated with prior methods (Figure 11). When compared against dehydrative Dean-Stark mediated conditions, the milder MIDA anhydride procedure provides higher isolated yields for sensitive boronic acids. A key advantage of the MIDA anhydride reagent is that it pre-loads one of the two dehydration steps required during the conversion of a boronic acid into a MIDA boronate. Moreover, excess MIDA anhydride acts as an internal desiccant promoting complexation through the generation of MIDA as an insoluble precipitate.
Figure 11. Head-to-head comparison of Dean-Stark complexation (A) and MIDA anhydride complexation (B) in the synthesis of MIDA boronates from boronic acids
On the basis of this operationally simple protocol using
MIDA anhydride, a highly accessible method for creating MIDA boronates directly from boronic acids was developed (Figure 12).
17 Crude material from
MIDA anhydride-based reactions can be purified
via a centrifuge column containing silica gel using a sequence of loading, washing, and eluting. The high crystallinity of the MIDA boronates enabled isolation of the product
via precipitation directly from the eluent stream. The application of this procedure to a range of alkyl, aryl and heteroaryl boronic acids allowed a diverse range of MIDA boronates to be prepared without recourse to any standard synthetic manipulations such as rotary evaporation and aqueous work-up.
Figure 12. Operationally simple synthesis of MIDA boronates enabled by MIDA anhydride in tandem with centrifuge-based catch and release purification/precipitation
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Acetic anhydride: (108-24-7)
Acetone: (67-64-1)
5-Acetylthiophene-2-boronic acid: (206551-43-1)
Diethyl ether: (60-29-7)
Dioxane: 1,4-dioxane; (123-91-1)
Ethyl acetate: (141-78-6)
MgSO4: Magnesium sulfate; (7487-88-9)
MIDA: Methyliminodiacetic acid; (4408-64-4)
MIDA anhydride: 4-Methylmorpholine-2,6-dione; (13480-36-9)
Pyridine: (110-86-1)
Toluene: (108-88-3)

|
Dr. Marty Burke is the May and Ving Lee Professor for Chemical Innovation, and Associate Dean for Research, Carle Illinois College of Medicine. Dr. Burke's group has pioneered the field of molecular prosthetics, and the development of an automated Lego-like platform for democratizing small molecule synthesis. Dr. Burke has received a number of honors and awards, including the Nobel Laureate Signature Award for Graduate Education in Chemistry from the American Chemical Society and has garnered praise for excellence in teaching at both the undergraduate and graduate levels. Dr. Burke, is an Academic Founder/Co-Founder of Revolution Medicines, Ambys Medicines, Sfunga Therapeutics, and cystetic Medicines, and led the SHIELD team at The University of Illinois, which is performing fast and frequent testing for all of our staff, faculty, and students. |

|
Daniel Blair completed his MSci in Chemistry at The University of Bristol (UK) in 2011. He continued his studies at Bristol undertaking a Ph.D. in Organic Chemistry under the guidance of Prof. Varinder K. Aggarwal, FRS focusing on stereoselective organolithium and organoboron chemistry, graduating in 2016. Daniel is currently pursuing postdoctoral research in the Burke laboratory and was an Illini 4000 Fellow of the Damon-Runyon Cancer Research Foundation. |

|
Peng-Jui (Ruby) Chen grew up in Taiwan, and then moved to Champaign, Illinois to attend University of Illinois at Urbana-Champaign as a chemistry major with a minor in computer science. She is currently performing undergraduate research under the supervision of Professor Martin Burke, focusing on understanding the metal mobilization activity of the natural product hinokitiol. |

|
Aidan Kelly grew up in Darien, IL, and received a Bachelor of Science in Biochemistry from the University of Illinois at Urbana-Champaign. His undergraduate research in the lab of Prof. Martin Burke focused on the development of simple and efficient MIDA boronate complexation methodology. In 2020, he moved to the Broad Institute of MIT and Harvard, where he currently works in the Biophysics group at the Center for Development of Therapeutics. |

|
Jack Hayward Cooke recieved an MChem in Chemistry from the University of Oxford in 2019, working on asymmetric catalysis with Prof. Darren Dixon. He is now pursuing Ph.D. studies in Chemistry under the guidance of Prof. Richmond Sarpong at the University of California, Berkeley, working on natural product total synthesis. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved