Org. Synth. 2022, 99, 363-380
DOI: 10.15227/orgsyn.099.0363
Preparation of 1,2:5,6-Di-O-cyclohexylidene-D-mannitol and 2,3-Cyclohexylidene-D-glyceraldehyde
Submitted by N. R. Dhatrak,
1* A. B. Jagtap, and T. N. Shinde
Checked by Albert Kruger, Nicholas Hafeman, and Seble Wagaw
1. Procedure (Note 1)
A. 1,2:5,6-Di-O-Cyclohexylidine-D-mannitol (1). An oven-dried round-bottomed flask (250 mL) (Note 2) containing a Teflon-coated magnetic bar (3 cm bar shape) is charged with DMSO (17 mL) (Note 3). D-Mannitol (98%) (12 g, 65.9 mmol, 1 equiv) (Note 4) is added to the flask in small lots while stirring at 25-30 °C. A Teflon stopper is used when not adding reagents. The resulting turbid solution is stirred for the next 5 min. Cyclohexanone (15 mL, 145 mmol, 2.2 equiv) and triethyl orthoformate (3.3 mL, 19.8 mmol, 0.3 equiv) are each added in one portion by syringe with an interval of 2 min (Note 5). Boron trifluoride etherate solution (0.7 mL, 5.7 mmol, 0.09 equiv) is then added to the flask with a glass syringe (Notes 5, 6, and 7). The colorless, turbid solution is stirred at room temperature (25-30 °C) for 24 h. After this time, the turbidity dissipates, and a clear solution results at the end of the reaction. The reaction progress is monitored by TLC (Note 8) (Figure 1).

Figure 1. A) The reaction is started; B) After completion of the reaction; C) TLC at work up (photos provided by submitters)
The reaction mixture is then neutralized by the slow addition of a saturated aqueous solution of sodium bicarbonate (5 g in x 50 mL distilled water) while stirring at 20 °C. A small amount of heat and CO2 gas are liberated during the neutralization. Distilled water (150 mL) (Note 9) is added to this solution, and stirring is continued for 5 min, after which the stirring is stopped, and the flask is left undisturbed for 30 min. During this time, a white precipitate forms in the flask.
Figure 2. A) The product solidifies inside the flask; B) The solid suspended and lumps are crushed under distilled water (photos provided by submitters)
The contents of the flask are filtered through a Büchner funnel (Note 10), and the filter cake is transferred to a 500 mL beaker containing 400 mL of distilled water (Figure 2b). The lumps of solid are crushed into smaller pieces, and the solid is allowed to settle to the bottom of the beaker. The suspension is then filtered again using a Büchner funnel under a vacuum (90 mmHg). The filter cake is again suspended in distilled water (400 mL), and the solid is recovered again by filtration to provide 22.4 grams of solid.
Figure 3. A) Product recovered by filtration; B) Solid after filtration (photos provided by submitters)
The separated solid is added to a 250 mL Erlenmeyer flask and is dissolved in ethyl acetate (170 mL), which results in a homogeneous solution. The ethyl acetate layer is dried over anhydrous sodium sulfate (20 g) while stirring for 30 min. The solution is then filtered through a plug of cotton directly into a 500 mL round-bottomed flask. The filtrate is then concentrated using a rotary evaporator (30 °C, 35 mmHg) to provide a viscous liquid, which upon standing converts into a wax-like solid (Figure 4).
Figure 4. Crude product before crystallization (photo provided by submitters)
Petroleum ether (400 mL) (Note 11) is added to this flask, and the flask is equipped with a condenser connected to a nitrogen source and heated to reflux using a heating mantel until all the solid dissolves.
Figure 5. Crude product dissolved in petroleum ether (photos provided by submitters)
The flask is then allowed to cool to room temperature (25 °C) and is left undisturbed for crystallization over 16 h. Soft crystals permeate the entire flask, and the liquid has the appearance of a soft white mass after 24 h (Figures 6A and 6B).
Figure 6. A) Partly crystallized product B) Fully crystallized product (photos provided by submitters)
This product is then loosened inside the flask using a long stainless-steel spatula and is filtered using a Büchner funnel under vacuum (90 mmHg). This provides a soft cake (Figure 7A), which is cut into pieces while the cake is wet (Figure 7B). The pieces of the cake are then transferred back into the 500 mL round-bottomed flask. The product is suspended in petroleum ether (400 mL), and the flask is heated to 75 °C to effect dissolution. The flask was allowed to cool to room temperature and left undisturbed for 16 h for crystallization to occur. The product is loosened inside the flask using a long stainless-steel spatula and is filtered using a Büchner funnel under vacuum (90 mmHg) (Note 12) .
Figure 7. A and B) Product recovered after vacuum filtration; C) Powdered product (photos provided by submitters)
The cake is then cut into small pieces and is dried in a warm oven (50 °C) for 16 h. The product is dried using a vacuum oven at 50 °C (Note 13). The product is stored in an air-tight bottle at room temperature (Note 14). The process yielded 10.89 g (48%) of compound 1 (Notes 15 and 16). The purity of the compound (1) is analyzed by quantitative 1H NMR and found to be 97.8% pure (Notes 17 and 18).
B. 2,3-Cyclohexylidene-D-glyceraldehyde (2). A 250-mL round-bottomed flask is charged with sodium periodate (3.25 g, 15.19 mmol, 1.3 equiv) and distilled water (30 mL) under ambient atmosphere. A Teflon stirring bar (2 cm bar-shaped) is added and the solution is stirred at ambient temperature until all the NaIO4 is dissolved. Diethyl ether (50 mL) is added to the flask, and 1,2:5,6-di-O-cyclohexylidene-D-mannitol (4 g, 11.68 mmol, 1 equiv) is added to the flask while stirring, to obtain a colorless biphasic mixture (Figure 8). Tetrabutylammonium bromide (160 mg, 0.5 mmol, 0.04 equiv) is added to the flask (Note 19), the flask is sealed with a Teflon stopper, and the resulting suspension is stirred for 3 h at room temperature.
Figure 8. A) Reaction before addition of Bu4NBr; B) Reaction after addition of Bu4NBr; C) TLC at the completion of the reaction (photos provided by submitters)
The reaction progress is monitored by TLC, and the product is visualized with 2,4-DNP solution (Note 20) (Figure 8C). After the reaction is judged complete, the reaction mixture is transferred to a 250 mL separatory funnel and the upper organic layer is collected in a 250 mL conical flask. The lower aqueous layer is extracted with diethyl ether (3 x 20 mL). The combined ether extracts are dried over anhydrous sodium sulfate (10 g). The dried organic layer is then filtered into a 250 mL round-bottomed flask through a cotton plug fitted in the stem of a glass funnel.
Figure 9. A) Short path distillation under vacuum (Note 22); B) Distilled compound 2 (photos provided by submitters)
The organic layer is concentrated using a rotary evaporator (30 °C, 35 mmHg) (Note 21). Purification is performed by distillation using a short path distillation apparatus at 57-61 °C (0.5-1.0 mmHg) (Note 22) (Figure 9A) to yield 1.91 grams (49% yield) of a clear liquid (Figure 9B) (Notes 23 and 24). The purity of the distilled compound (2) is determined to be 99.1% by quantitative 1H NMR (Note 25).
2. Notes
1. Prior to performing each reaction, thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
DMSO,
D-mannitol,
cyclohexanone,
triethyl orthoformate,
boron trifluoride etherate,
sodium bicarbonate,
ethyl acetate, petroleum ether,
sodium periodate,
diethyl ether,
tetrabutylammonium bromide,
2,4-DNP solution, anhydrous
sodium sulfate,
chlorobenzaldehyde, and
CDCl3.
2. The reaction uses
DMSO as a solvent and should be handled carefully. It is suggested to use nitrile gloves when performing the entire reaction sequence. Glass stoppers are not recommended as the reaction forms a pasty mass and
BF3 gas can irreversibly lock the glass stoppers. It is suggested to use polypropylene or Teflon stoppers instead of glass stoppers.
3.
DMSO (HPLC Grade, obtained from Sigma-Aldrich) was used without any further purification. Gloves are worn when handling
DMSO and also while handling the aqueous solutions of
DMSO during aqueous workups.
4.
D-Mannitol (≥98%) was purchased from Sigma Aldrich.
5.
Triethyl orthoformate (98% anhydrous),
cyclohexanone (≥99.5%), and
Boron trifluoride etherate solution (44-51%
BF3) are purchased from Sigma-Aldrich. The reaction requires 2 equiv of
Cyclohexanone, but an excess of 0.2 equivalent is used, which was determined to assist in the optimization of the yield.
6.
Boron trifluoride etherate solution gives off fumes, and the bottle should be cooled before opening. The opening of this bottle and the addition of the contents should be carried out in a well-ventilated fume hood.
7. The glass syringe used for the addition of
BF3•OEt2 solution must be washed immediately with water, then with dil.
HCl and rinsed with water again.
8. The reaction progress can be monitored by TLC on silica using 35%
ethyl acetate in petroleum ether. The reactant and the product both are visualized by staining the TLC in
2,4-DNP solution. The product spot appears at R
f = 0.34.
9. Distilled water is used to avoid inorganic impurities. Normal tap water can also be used in this step.
10. The reaction mixture is diluted with water and contains a large quantity of
DMSO. This aqueous solution should be processed as per prudent practices before disposal.
11. Petroleum ether (60-80 °C, Sigma Aldrich) was used for crystallization. It was distilled with a rotary evaporator before use and was dried over 50 g of anhydrous
sodium sulfate for an hour while stirring.
12. The cake obtained after the second crystallization followed by filtration is free from the unreacted
cyclohexanone.
13. The submitters report that the product is dried using a rotary evaporator for 30 min (5 mmHg, 80 °C). The dried product (Figure 7C) is transferred to a 250 mL round-bottomed flask, and a cotton plug is pushed into the neck of the flask, which prevents particles of product from going inside the vapor tube of the rotary evaporator.
14. The product is stable and is not hygroscopic. It is stored under nitrogen to prevent air oxidation. The product recovered after the second crystallization is sufficiently pure for the next reaction, however, if required, it can be further purified by column chromatography.
15. A second reaction on identical scale provided 9.20 g (41%) of the same product.
16. Characteristics of the compound (
1): mp 105-106 °C; IR (ATR) 3306 (broad), 2931, 2855, 1106, 1043, 921 cm
-1.
1H NMR
pdf (400 MHz,
CDCl3) δ: 4.19 (q,
J = 6.2 Hz, 2H), 4.11 (dd,
J = 8.5, 6.3 Hz, 2H), 3.96 (dd,
J = 8.5, 5.5 Hz, 2H), 3.74 (t,
J = 6.4 Hz, 2H), 2.71 (d,
J = 6.5 Hz, 2H), 1.67 - 1.53 (m, 16H), 1.43 - 1.34 (m, 4H). .
13C NMR
pdf (100 MHz,
CDCl3) δ: 110.2, 76.1, 71.5, 66.6, 36.6, 34.8, 25.2, 24.2, 23.9. [α]
D20 = +5.95. HRMS, [M+Na], calculated for C
18H
30O
6Na, found to be 365.1938
17. The purity of the compound (
1) was found to be >95% by quantitative NMR
pdf analsyis using
1,3,5-trimethoxybenzene as the internal standard.
18. If material with higher purity is desired, the submitters report that the crystallized product can be further purified by silica gel column chromatography with column dimensions 600 x 40 mm, Silica gel 100-200 mesh 180g, and distilled
ethyl acetate and petroleum ether, gradient 3 to 15%. TLC (35%
ethyl acetate in petroleum ether) can be used to monitor the column eluate. An effective visualization reagent is
2,4-DNP. The product spot appears at R
f = 0.34.
19. The
tetrabutylammonium bromide (98%) was purchased from Sigma-Aldrich. The reaction mixture becomes turbid upon the addition of the
tetrabutylammonium bromide. This observation is normal for this reaction. TBAI is not suitable for the reaction as it releases iodine in the reaction.
20. TLC is performed with 15%
ethyl acetate in petroleum ether, by developing (eluting) the same TLC plate 5 times. The product aldehyde and the starting diol both were found to show orange-colored spots when exposed to
2,4-DNP stain. Compound (
1) shows a light orange spot in while the product aldehyde (
2) shows a dark orange spot. The compound (starting material) spot disappears on heating the TLC.
21. The crude product aldehyde (
2) can be used directly without any further purification.
22. Chromatographically purified samples (silica and alumina) show significantly lower optical rotation, which suggests that chromatographic purification is best avoided. The aldehyde (
2) obtained after the distillation shows significantly higher optical rotation (+80.2) than reported earlier (+61.2,
2 +61.5,
3 and +50 to 70
4). The submitters utilized a Hickman still (illustrated in Figure 9) in combination with the short path apparatus. The submitters reported that distillation between 90-110 °C is suitable for the distillation of the product. In addition, the submitters cautioned that distillation to dryness resulted in the generation of pale, yellow-colored impurities in the product.
23. Characteristics of compound (
2):
1H NMR
pdf (400 MHz,
CDCl3) δ: 9.71 (d,
J = 2.0 Hz, 1H), 4.37 (ddd,
J = 7.0, 4.6, 1.9 Hz, 1H), 4.15 (dd,
J = 8.8, 7.4 Hz, 1H), 4.08 (dd,
J = 8.8, 4.7 Hz, 1H), 1.73 - 1.53 (m, 10 H),
13C NMR
pdf (100 MHz,
CDCl3) δ: 202.1, 111.9, 79.6, 65.2, 35.9, 34.6, 25.0, 23.9, 23.8. IR (ATR) 2933.4, 2858.9, 1733.2, 1449.9, 1367.9, 1334.4, 1282.2, 1162.9, 1092.1, 920.7, 846.1, 775.3 cm
-1. [α]
D20 +80.2° (c 4.81, CHCl
3); HRMS [M+H] calculated for C
9H
15O
3, was found to be 171.1015
24. In a second reaction on the same scale the checkers obtained 2.35 g (59%) of the target product.
25. The checkers assessed purity to be 99.1 % by Q NMR
pdf using
1,3,5-trimethoxybenzene (11.2 mg) as the internal standard with
2,3-cyclohexylidene-D-glyceraldehyde (41.3 mg).
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
1,2:5,6-Di-O-cyclohexylidene-D-mannitol (
1) is one of the protected forms of
D-mannitol.
2 Both terminal 1,2-diol functions are protected with a cyclohexylidene group, and the central diol functionality is available for chemical transformations and is generally subjected to oxidative cleavage.
2 This diol function is readily cleaved on reaction with sodium meta periodate (
NaIO4) to obtain an optically pure protected glyceraldehyde (
2,3-O-cyclohexylidene-D-glyceraldehyde).
2 The aldehyde has a chiral carbon of known stereochemistry
(R) and has been used as a chiral starting material in many synthetic efforts. Aldehyde
2 is freshly prepared before use because of its relative instability; therefore,
1,2:5,6-di-O-cyclohexylidene-D-mannitol (
1) presents as a stable and ready resource of
2,3-O-cyclohexylidene-D-glyceraldehyde (
2).
1,2:5,6-Di-O-cyclohexylidene-D-mannitol (
1) is readily cleaved into two molar equivalents of aldehyde (
2) using
NaIO4 in
diethyl ether/water system with added
tetrabutylammonium bromide as phase-transfer catalyst
2 or NaIO
4 adsorbed on silica gel in DCM.
2 This yields two molar equivalents of optically pure
2,3-O-cyclohexylidene-D-glyceraldehyde (
2).
2 The cyclohexyl protection can be removed easily by aqueous conc.
HCl in methanol at room temperature.
5
Scheme 1. a) NaIO4, H2O, /Et2O 12 h, rt or NaIO4/Silica, DCM, 7 h, rt
Aldehyde (
2), upon reaction with vinyl magnesium bromide, yields alcohol (
3) and has been used for the synthesis of a novel macrocyclic peptide.
6Scheme 2. a) Vinyl magnesium bromide, THF, 0 °C, rt, 6 h, then aq. NH4Cl
Aldehyde
2 on reaction with allyl bromide in the presence of Zn metal, yields alcohol
4a with a diastereomeric excess of 94%.
7Scheme 3. a) Allyl bromide, Zn, aq. NH4Cl, 0 °C, 4 h, rt
The syn alcohol (
4b) is synthesized by oxidation of compound
4a followed by
K-Selectride
TM-mediated reduction. Thus, the required stereochemistry of the hydroxyl group can be achieved.
8 Alcohol
4b has been used for the stereoselective synthesis of C
4-C
18 fragment of arenicolide-A.
8Scheme 4. a) allyl magnesium bromide, THF, 0 ºC, b) PCC, DCM, 3h, rt, c) K-SelectrideTM
Allyl chloromethyl ether
9 on reaction with triphenylphosphine yields allyloxymethyltriphenylphosphonium chloride salt
10 which on reaction with an aldehyde
2 yields corresponding allyl vinyl ether
7 as an inseparable mixture of
cis and
trans isomers.
5 Allyl vinyl ethers so generated are excellent substrates for Claisen rearrangement.
5 On Claisen rearrangement, compound
7 is converted into a new aldehyde
8 which on reduction provides a chromatographically separable mixture of diastereomeric alcohols
(9a,b). Both the diastereomers (
9a,b) have been separated by simple column chromatography and the stereochemistry of new chiral carbon is established by converting them into known compounds.
5 These alcohols (
9a,b) have been used as chiral auxiliaries in the syntheses carbahexofuranoses
5, optically pure isomers of
bis-THF derivative
11 (+)-isofagomine and bis THF.
12
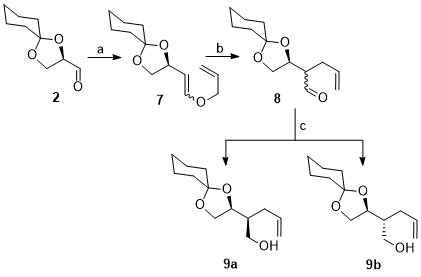
Scheme 5. a) Allyloxymethyltriphenylphosphonium chloride, t-BuONa, THF, 0 °C, 2 h, b) Xylene, reflux 24 h, c) NaBH4, MeOH, rt, followed by chromatographic separation
Diene systems suitable for ring closing metathesis
(10 and
12) have been prepared from aldehyde (
8). Nucleophilic addition of vinyl and allyl groups to aldehyde
8 yields suitable substrates for ring closing metatheses. This method has afforded cyclopentenol
5 11 and cyclohexenone
13 13 derivatives and has been used for stereospecific syntheses.
Scheme 6. Vinyl magnesium bromide, THF, 0 °C, 2 h, then aq. NH4Cl, b) Ino, allyl bromide, THF/H2O (30%), rt, 6 h, c) Grubbs catalyst IInd Gen (5 mol%), DCM, rt, 6 h
1,2:5,6-Di-O-cyclohexylidene-D-mannitol (
1) is a versatile and useful reagent for many organic syntheses. Compound
1 is comparable with
1,2:5,6-diisopropylidene-D-mannitol.
14 The diol protection in
1 is more challenging than the corresponding diacetonide (1,2:5,6-Di-
O-isopropylidene-D-mannitol. Besides, it also offers an appropriate steric hindrance for asymmetric syntheses due to the bulky cyclohexyl groups. The aldehyde (
2) has numerous applications in organic synthesis
15 and
1,2:5,6-Di-O-cyclohexylidene-D-mannitol serves as a stable resource for
2. The purified aldehyde (
2) shows significantly higher optical rotation (+80.2) than the earlier reports (+61.2,
2 +61.5,
3 and +50 to 70
4).
Appendix
Chemical Abstracts Nomenclature (Registry Number)
D-mannitol; (69-65-8)
Boron trifluoride etherate; (109-63-7)
Triethyl orthoformate; (122-51-0)
Cyclohexanone; (108-94-1)
Dimethyl sulfoxide; (67-68-5)
Sodium sulfate; (7757-82-6)
Sodium periodate; (7790-28-5)
Tetrabutylammonium bromide (1643-19-2)

|
Dr. Dhatrak N. R. was born in India. He did his M.Sc. Chemistry from Government Institute of Science and Humanities, Amravati, and had completed a Ph.D. in organic synthesis from Savitribai Phule Pune (formerly known as the University of Pune). He taught chemistry at the graduate level at Anand Niketan College, Anandwan, a premier institute founded by renowned social worker Baba Amte for six years. Currently, he is working as an Associate Professor of chemistry, at the Department of Chemistry, Savitribai Phule Pune University. His research areas are the total synthesis of natural products, design, and development of novel instruments for chemistry. |

|
Anuja B. Jagtap. was born in India. She did her M.Sc. Chemistry from the Department of Chemistry, Savitribai Phule Pune University, Pune, and currently working for her Ph.D. degree in Organic synthesis under the guidance of Dr. Dhatrak N. R. at the Department of Chemistry, Savitribai Phule Pune University, Pune (formerly known as the University of Pune). Her research areas are organic synthesis and synthesis of nanomaterials. |

|
Trupti N. Shinde. was born in India. She did her M.Sc. Chemistry from Abeda Inamdar College Pune, and currently working for her Ph.D. degree in Organic synthesis under the guidance of Dr. Dhatrak N. R. at the Department of Chemistry, Savitribai Phule Pune University, Pune (formerly known as the University of Pune). Her research areas are organic synthesis and total synthesis of natural products. |

|
Albert Kruger has been in AbbVie Process Chemistry for 22 years. He received his B.S. from the University of Nebraska (Lincoln) and his Ph.D. from the University of Wisconsin (Madison). While at Wisconsin, he studied enantioselective protonation of enolates in the labs of Professor Ed Vedejs. |

|
Nick Hafeman is a Senior Scientist in Process Chemistry at AbbVie. He received his B.S. from the University of Illinois at Chicago in 2016, where he performed research under the guidance of Prof. Duncan Wardrop. He then joined the laboratory of Prof. Brian Stoltz at Caltech, where he studied the total synthesis of complex terpenoid natural products, earning a PhD in 2022. Following his graduate studies, he joined the process team at AbbVie in July, 2022. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved