Org. Synth. 1993, 71, 146
DOI: 10.15227/orgsyn.071.0146
ETHYL 1-NAPHTHYLACETATE: ESTER HOMOLOGATION VIA YNOLATE ANIONS
[1-Naphthaleneacetic acid, ethyl ester]
Submitted by Rajarathnam E. Reddy and Conrad J. Kowalski
1.
Checked by Chang Y. Hong and Larry E. Overman.
1. Procedure
A. Preparation of lithium 2,2,6,6-tetramethylpiperidide (LiTMP). An oven-dried, 500-mL, round-bottomed flask, equipped with a magnetic stirring bar, nitrogen inlet, and septum, is filled with a nitrogen atmosphere and charged via syringe with 40.5 mL (33.9 g, 0.240 mol) of 2,2,6,6-tetramethylpiperidine (Note 1) and 250 mL of anhydrous tetrahydrofuran (Note 2). The rapidly stirred solution is cooled with an ice bath and 88.0 mL (0.220 mol) of 2.5 M butyllithium in hexanes (Note 3) is added via syringe over a 20-min period. This solution of lithium 2,2,6,6-tetramethylpiperidide is stirred with continued ice bath cooling for a period of about 30 min while the next reaction is readied.
B. In situ lithiodibromomethane preparation/addition. An oven-dried, 2-L, round-bottomed flask with three vertical necks, containing 20.0 g (0.100 mol) of ethyl 1-naphthoate (Note 4), is equipped with a 2-1/2 × ¾-inch, egg-shaped magnetic stirring bar (Note 5), a −100°C to +50°C thermometer (Note 6) and a rubber septum. The apparatus is flushed with nitrogen and a nitrogen atmosphere is maintained throughout the reaction using a nitrogen bubbler system that allows pressure release. The flask is charged through the septum (via syringe) with 200 mL of anhydrous tetrahydrofuran and 15.5 mL (38.3 g, 0.220 mol) of dibromomethane (Note 1). The thermometer (in a side neck) is adjusted so that the bulb is submerged when the liquid is stirred. The flask is immersed almost to the necks in a dry ice-acetone bath using about 2 L of acetone, and the contents are stirred vigorously for good heat transfer.
The lithium tetramethylpiperidide solution from Part A is cooled with a dry ice-acetone bath. When the solution of ethyl 1-naphthoate and dibromomethane has cooled to −74°C or below (internal solution temperature), addition of the dry ice-acetone cooled solution of lithium tetramethylpiperidide is begun. Addition is made via a double-ended (16 gauge) needle over a 40 to 50-min period using a slight positive nitrogen pressure in the 500-mL flask (Note 7). During this time, the addition rate is slowed or stopped as needed to maintain the reaction temperature below −67°C.
C. Preparation of lithium hexamethyldisilazide(LiHMDS)/lithium ethoxide. As the addition of Part B above is progressing, an oven-dried, 500-mL, round-bottomed flask, equipped with a magnetic stirring bar, nitrogen inlet, and septum, is filled with a nitrogen atmosphere and charged via syringe with 42.1 mL (32.2 g, 0.200 mol) of 1,1,1,3,3,3-hexamethyldisilazane (Note 1), 5.9 mL (4.6 g, 0.100 mol) of absolute ethanol, and 160 mL of anhydrous tetrahydrofuran. The rapidly stirred solution is cooled with an ice bath and 120 mL (0.300 mol) of 2.5 M butyllithium in hexanes is added via syringe over a 20-min period. The resulting solution of lithium hexamethyldisilazide and lithium ethoxide is stirred with continued cooling for a period of about 30 min and is then cooled with a dry ice-acetone bath (Note 8).
D. Base and butyllithium addition. Ten minutes after the addition of lithium tetramethylpiperidide to the main reaction is complete, the cold (ca. −70°C) solution of lithium hexamethyldisilazide and lithium ethoxide is added (via a double-ended needle) over a 15 to 20-min period, while maintaining the reaction mixture temperature below −65°C. Five minutes after the addition is complete, the dry ice-acetone cooling bath is removed and the reaction mixture is allowed to warm. When the internal temperature of the solution has reached −20°C (after about 15 min), the reaction mixture is again cooled with a dry ice-acetone bath (Note 9), (Note 10). When the internal reaction temperature has reached −70°C or below, 308 mL (0.400 mol) of 1.3 M sec-butyllithium in cyclohexane (Note 11) is added via syringe over a 30 to 40-min period so that the internal temperature is maintained below −60°C. Five minutes after the addition is complete, the dry ice-acetone cooling bath is removed and the reaction mixture is allowed to warm. After the internal temperature has gradually reached ca. −10°C (Note 12), the reaction mixture is warmed to 20°C with a ~25°C water bath. A solution of butyllithium (80 mL, 0.200 mol) in hexanes is then added using a syringe pump over a 30 to 40-min period while the internal temperature is maintained between 20–25°C (Note 13), (Note 14). The reaction mixture is then allowed to stir for 30 min at room temperature (Note 15).
E. Quench and purification. While the butyllithium addition is taking place, an acidic ethanol quench solution is prepared in a 3-L, two-necked, round-bottomed flask, equipped with a mechanical stirrer and a 250-mL, pressure-equalizing dropping funnel. The flask is charged with 1 L of absolute ethanol and the funnel with 250 mL of acetyl chloride. The ethanol is stirred rapidly and the flask is cooled with an ice bath as the acetyl chloride is added over a 30–40-min period and then the cooling bath is removed and stirring is continued for 20–30 min. After the main reaction mixture has been stirred for 30 min at room temperature, it is cooled with a dry ice-acetone bath. The acidic ethanol solution is cooled with an ice bath and the cold, main reaction mixture is quenched by addition (via a double-ended needle) into the rapidly stirred, cold, acidic ethanol solution over a 3 to 3.5 hr period (Note 16).
After the quench is complete, the mixture is diluted with 2 L of ether and washed in a separatory funnel with 1.25 L of aqueous 10% hydrochloric acid. The aqueous layer is reextracted with a 2-L portion of ether. The combined ethereal layers are washed with 1.25 L of saturated brine solution and dried over anhydrous magnesium sulfate. After filtration and removal of solvent using a rotary evaporator there remains 26 g of crude product as a reddish-brown oil. Distillation of this material under reduced pressure through an unpacked, 2-inch distillation apparatus affords 17.3 g (81%) of ethyl 1-naphthylacetate as a yellow liquid, (bp 134–138°C, 0.2 mm) (Note 17), (Note 18).
2. Notes
1.
2,2,6,6-Tetramethylpiperidine, dibromomethane (99%) and 1,1,1,3,3,3-hexamethyldisilazane (98%) were purchased from Aldrich Chemical Company, Inc., and used without further purification. Use of less hindered secondary amines (such as
diisopropylamine) in place of
tetramethylpiperidine results in lower yields because of the formation of carboxamide by-products.
2.
Reagent grade tetrahydrofuran was freshly distilled under
nitrogen from a purple solution of
sodium and
benzophenone.
3.
Solutions of
butyllithium in hexanes and sec-butyllithium in cyclohexane were purchased from the Aldrich Chemical Company, Inc. It is recommended that only freshly opened bottles or extremely well protected solutions be used as the presence of
lithium butoxide in partially decomposed bottles results in formation of the corresponding butyl ester as an undesired by-product.
4.
Ethyl 1-naphthoate was purchased from Lancaster Synthesis, Inc., P.O. Box 1000, Windham, NH 03087, and used without purification. The checkers found that the purity of the ester was 95% by capillary GC analysis.
5.
Good stirring is important to the success of this reaction, in order to provide efficient heat transfer during the addition of
lithium tetramethylpiperidide and
sec-butyllithium. The 2-1/2 × ¾ inch stirring bar used is available from Aldrich Chemical Company, Inc.
6.
The submitters recommend calibrating the thermometer to ca. −78°C when immersed in a dry ice-acetone bath. The thermometer used for these runs (Catalog #Z11,011-6) was purchased from Aldrich Chemical Company, Inc. Three different thermometers (two Fisher 15-035 and one Ertco X7048) were found that read from −86°C to −96°C when immersed in a dry ice-acetone bath! The checkers used a
digital thermometer, model 450-ATT, purchased from OMEGA Engineering, Inc., Stamford, CT 05907.
7.
At a very low temperature the
lithium tetramethylpiperidide solution may become too thick for smooth addition. If that occurs, the dry ice-acetone cooling bath is either partially or completely removed for a short period until addition can take place. The checkers warmed this mixture to −20°C to −10°C (whereupon it became a clear solution) prior to addition.
8.
While it is important for all ester substrates that
lithium hexamethyldisilazide be added before warming in order to avoid yield loss, the addition of
lithium ethoxide (LiOEt) is specific for the naphthyl ester and is not generally necessary (see Discussion). Thus for other esters the
ethanol can be omitted in this step and the amount of
butyllithium can be reduced to 0.20 mol.
9.
The reaction mixture turns from a yellow-orange color to a dark brown/black during the warming process, at ca. −35° to −25°C.
10.
It is helpful to withdraw about a 0.20-mL aliquot of the reaction mixture after the warming-recooling process, to quench it in about
1.5 mL of a 1:5 mixture of acetyl chloride and absolute ethanol, and then to add about
1 mL of ethyl acetate and 0.5 mL of water. The mixture is shaken and most of the organic layer is transferred via
pipette to a tube containing anhydrous
magnesium sulfate for drying. GC analysis of this material (at a convenient time) on a 50 m × 0.32 mm i.d. capillary column of cross-linked methylsilicone, at 40 psi of pressure for the
helium carrier gas, and at a temperature raised from 100°C to 300°C at a rate of 15°C/min should show no remaining starting ester (ret. time = 6.5 min); instead it should show dibromo ketone
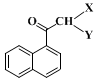
Ia (X = Br, Y = Br) at a ret. time = 9.0 min, monobromo ketone
Ib (X = H, Y = Br) at a ret. time = 7.7 min and monochloro ketone
Ic (X = H, Y = Cl) at a ret. time = 7.1 min (resulting from halogen exchange during the quench) in a ratio of about 3.9:4.2:1.
11.
Use of
butyllithium in place of
sec-butyllithium at this point results in formation of some α-butylated ester by-product later upon warming, while use of
sec-butyllithium in greater excess results in formation of methyl ketone
Id (X, Y = H) in the final product.
12.
CAUTION: A pressure buildup can occur on this scale if the reaction mixture is warmed from −78°C to −10°C too rapidly and if the nitrogen inlet/pressure release system has very narrow constrictions. The initial warming is best done without a bath and takes about 15 min.
13.
The internal temperature is maintained between 20°C to 25°C during the addition with a cold water bath.
14.
For non-aromatic esters, this addition can be carried out more conveniently by adding the
butyllithium over 5–10 min (without cooling) once the internal temperature has reached −20°C, and then warming to room temperature using a 20–25°C bath. For
ethyl 1-naphthoate, however, addition of
butyllithium at a lower temperature, or too quickly at a higher temperature, results in formation of ca. 3.2% of methyl ketone
Id (X = Y = H). This lowers the yield and this compound is difficult to separate from the product.
15.
It is wise to remove, quench, and analyze an aliquot from the reaction (as described in
(Note 10). At this point there should be no dibromo ketone
Ia (X, Y = Br, ret. time = 9.0 min) or monobromo ketone (X = H, Y = Br, ret time = 7.7 min), but instead a strong peak for product at a ret. time = 6.8 min. If any monobromo or dibromo ketone is present in the 30-min aliquot, a longer time should be allowed for stirring at room temperature and/or slightly more
butyllithium should be added.
16.
It is important to quench the reaction
intothe acidic
ethanol. If the quench solution is added to the reaction mixture, very little product is obtained.
17.
The pot material in this distillation is rather viscous and tends to bump, so good stirring and extra care should be employed to avoid this problem. A low-boiling fraction (0.67–0.78 g, 95–133°C, 0.2 mm) is collected that is mostly a mixture of hydrocarbons and
ethyl 1-naphthylacetate (ca. 1% yield). More than 90% of the product distils at 134–138°C, 0.2 mm. For the final few drops of product the bath temperature must be raised by 15–20°C, resulting in a slight rise in product boiling point; however, all the remaining distillate obtained is desired product.
18.
G/C analysis (as in
(Note 10)) of the final product, shows a single peak with retention time of 6.8 min. This material has the following spectral properties: IR (neat) cm
−1: 2981, 1730, 1598, 1512, 1445, 1174, 792, 780;
1H NMR (CDCl
3) δ: 1.19 (t, 3 H, J = 7.0), 4.04 (s, 2 H), 4.13 (q, 2 H, J = 7.1), 7.39–7.51 (m, 4 H), 7.76 (dd, 1 H, J = 2.4, 7.1), 7.84 (d, 1 H, J = 7.9), 7.98 (d, 1 H, J = 8.3).
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
This procedure illustrates a general method for the direct homologation of common esters, that represents a convenient, safer alternative to the classical Arndt-Eistert approach.
Ethyl 1-naphthylacetate was selected for this demonstration since its preparation via the classical procedure has appeared previously in
Organic Syntheses,
2 thus offering a direct comparison. In the earlier work,
1-naphthoyl chloride was prepared from the corresponding acid and converted using
diazomethane to
1-(diazoacetyl)naphthalene. Preparation of multigram quantities of hazardous
diazomethane was required, and the
1-(diazoacetyl)naphthalene produced was a severe skin irritant that had to be recrystallized in order to achieve the desired overall yield. Freshly prepared
silver benzoate was then used to catalyze the Wolff rearrangement, providing the final
ethyl 1-naphthylacetate in
78% overall yield (from the acid chloride). A modified Arndt-Eistert procedure using
trimethylsilyldiazomethane has also been used to effect transformation of
1-naphthoyl chloride to
ethyl 1-naphthylacetate in
66% yield on a 3-mmol scale.
3The current procedure starts with the ester,
ethyl 1-naphthoate, and converts it into homologated product in
81% yield without isolation of any intermediates. Safe, commercially available materials are employed. Based on a recently published variation
4 of our original homologation methods,
5 it represents a general procedure applicable to esters
1 bearing aryl, alkenyl, alkynyl and primary, secondary or tertiary alkyl attachments R. Yields range from 67–90% (on a 25-mmol scale) and like the Arndt-Eistert sequence this procedure affords retention of stereochemistry for the attachment R. It is also worth noting that this chemistry proceeds through the intermediacy of ynolate anions
5 that are useful for other processes,
6 7 including the preparation of silyloxyacetylenes.
8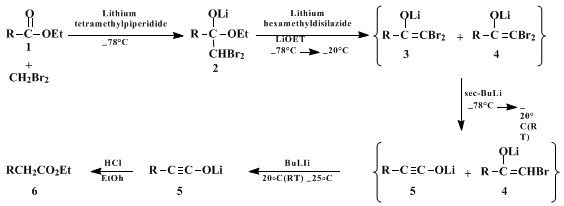
Parts A and B of the procedure correspond to preparation of
lithium tetramethylpiperidide, and its use in the in situ preparation
9 and addition of
dibromomethyllithium to the ester
1 producing tetrahedral intermediate
2. In Part C a mixture of
lithium hexamethyldisilazide and
lithium ethoxide is prepared for addition in Part D to the solution of
2. The silazide base serves to deprotonate the mono and dibromo ketones that are formed on initial warming of the reaction to −20°C, thus protecting them as the enolate anions
4 and
3. Addition of the
sec-butyllithium in Part D effects the key metal-halogen exchange/rearrangement of
3 at −78°C, converting it to the ynolate anion
5 (and producing
sec-butyl bromide that does not alkylate the ynolate anion on subsequent warming). Addition of
butyllithium, also in Part D, provides a source of base (regenerated
lithium tetramethylpiperidide) to deprotonate enolate
4 at room temperature and initiate rearrangement to ynolate
5. The Part E quench then affords final product
6.
Two modifications were made to our general procedure,
4 specifically to suit the
ethyl 1-naphthoate case in this preparation, that are not necessary for most other compounds. For most ynolate anions,
5, the inverse quench into acidic
ethanol described here is sufficient to eliminate significant formation of dimeric products resulting from ynolate anion reaction with the ketene intermediate. In the
naphthalene case, however, about 3% of dimeric material (which complicates the distillation) is formed, possibly because of tighter aggregation of the ynolate anions via π-stacking. This was eliminated by adding an equivalent of
lithium ethoxide (Parts C and D) to provide more ethoxide as an internal nucleophile/aggregate companion to trap effectively all the ketene formed in the quench. This ethoxide is not generally required for other substrates
(Note 8).
It was also found for the naphthalene case (and to a lesser extent for phenyl), that small amounts of metal-halogen exchange can take place between bromoenolate 4 and butyllithium at room temperature, affording about 3% of acetonaphthone after quench. Thus the butyllithium addition was carried out slowly enough at room temperature so that no buildup of butyllithium would occur; under these conditions, the lithium tetramethylpiperidide deprotonation of 4 was occurring sufficiently fast to generate tetramethylpiperidine and to protonate the butyllithium as it was being added. For non-aromatic R groups on 4, this competitive metal-halogen exchange was not a problem and the butyllithium need not be added slowly at room temperature (Note 14).
Ethyl 1-naphthoate thus represents one of the more difficult homologation cases, requiring the two modifications above. For most other compounds these are unnecessary and the procedure can be simplified as noted. On untried compounds one should always test the chemistry first on a small (2 mmol) scale, monitoring the quenched aliquots (as in
(Note 10) and
(Note 15)) to ensure that minor adjustments to the stoichiometry are not needed (e.g., a larger excess of
dibromomethyllithium in the first step to consume all the starting ester and/or more
butyllithium in the final step to deprotonate/rearrange all the bromoenolate
4). No such changes have been required for any of the esters that we have investigated to date,
4 however, indicating that this procedure represents a quite general method for the direct homologation of common esters.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Lithium 2,2,6,6-tetramethylpiperidide
brine
Lithium hexamethyldisilazide
lithium 2,2,6,6-tetramethylpiperidide (LiTMP)
ethanol (64-17-5)
hydrochloric acid (7647-01-0)
ethyl acetate (141-78-6)
ether (60-29-7)
acetyl chloride (75-36-5)
sec-Butyl bromide (78-76-2)
nitrogen (7727-37-9)
cyclohexane (110-82-7)
acetone (67-64-1)
Benzophenone (119-61-9)
sodium (13966-32-0)
Naphthalene (91-20-3)
dibromomethane (74-95-3)
magnesium sulfate (7487-88-9)
Diazomethane (334-88-3)
ethyl 1-naphthoate (3007-97-4)
1-naphthoyl chloride (879-18-5)
acetonaphthone (941-98-0)
butyllithium (109-72-8)
Tetrahydrofuran (109-99-9)
helium (7440-59-7)
lithium ethoxide
diisopropylamine (108-18-9)
2,2,6,6-tetramethylpiperidine (768-66-1)
Ethyl 1-naphthylacetate (3121-70-8)
1-Naphthaleneacetic acid, ethyl ester (2122-70-5)
1-(Diazoacetyl)naphthalene (4372-76-3)
silver benzoate (532-31-0)
sec-butyllithium (598-30-1)
1,1,1,3,3,3-hexamethyldisilazane (999-97-3)
lithium tetramethylpiperidide
lithiodibromomethane,
Dibromomethyllithium
tetramethylpiperidine
lithium butoxide (2698-11-5)
lithium dicyclohexylamide
Trimethylsilyldiazomethane (18107-18-1)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved