Org. Synth. 2009, 86, 298
DOI: 10.15227/orgsyn.086.0298
TETRAKIS(DIMETHYLAMINO)ALLENE
Submitted by Alois Fürstner, Manuel Alcarazo, and Helga Krause
1.
Checked by Peter Wipf and Carl Deutsch
2.
1. Procedure
Caution! This procedure should be carried out in a well ventilated hood because of the evolution of HCl gas (step A), dimethylamine (step B) and NH3 (step C), which are corrosive, noxious and irritant agents.
A. 1,3-Dichloro-1,3-bis(dimethylamino)propenium chloride. A 250-mL, two-necked, round-bottomed, flame-dried flask equipped with a Teflon-coated stir bar (4 cm), a glass stopper, and a reflux condenser (20 cm) fitted with an argon inlet is charged with dichloromethylene-dimethyliminium chloride (15.50 g, 95.41 mmol, 2 equiv) (Note 1), dimethylacetamide (4.15 g, 47.71 mmol, 1 equiv) (Note 1) and dichloromethane (100 mL) (Note 2). The suspension is heated at reflux for 3 h in an oil bath at 47 °C. During this time, a constant evolution of HCl gas is observed while all solid materials dissolve and the solution develops an intense yellow to orange-red color. After reaching ambient temperature, the reflux condenser is replaced by a short path distillation head (10 cm) and all volatile materials are distilled off under reduced pressure (10 mmHg, bath temperature ca. 40 °C) to give 1,3-dichloro-1,3-bis(dimethylamino)-propenium chloride as bright yellow, air-sensitive crystals that are dried in vacuo (10−3 mmHg). The crude material is used in the next step without further purification (10.68 g, 97%) (Note 3).
B. 1,1,3,3-Tetrakis(dimethylamino)propenium tetrafluoroborate. A 250-mL, two-necked, round-bottomed, flame-dried flask equipped with a Teflon-coated stir bar (4 cm), an argon inlet, and a glass stopper is charged under argon with crude 1,3-dichloro-1,3-bis(dimethylamino)propenium chloride (10.4 g, 45 mmol, 1 equiv) (Note 3). The flask is immersed into a dry ice/acetone cooling bath at −78 ºC.
The volume of 23 mL is clearly marked on the outside of a one-necked Schlenk tube (50 mL, 15 cm length) (Note 4) equipped with a glass stopper and connected to the argon/vacuum line via its side arm. The flask is flame dried in vacuo (0.3 mmHg), back-filled with argon, and allowed to reach ambient temperature. The glass stopper is replaced by a gas inlet and the flask is immersed into a dry ice/acetone bath. Dimethylamine (ca. 23-26 mL, 350-390 mmol, 8 equiv) (Note 1) is then gently condensed from a commercial cylinder into the flask (Note 5). By applying slight argon overpressure, the condensed dimethylamine is rapidly (< 1 min) transferred via a teflon tube (2 mm diameter) into the round-bottomed flask containing the chilled 1,3-dichloro-1,3-bis(dimethylamino)propenium chloride. The resulting mixture is vigorously stirred for 1 h at −78 °C and for 1 h at −20 °C using an external thermometer in a dry ice/acetone bath. During this time, the yellow solid disappears and a white precipitate is formed. The suspension is then warmed to room temperature, allowing the excess dimethylamine to evaporate through the argon bubbler (Note 6). The remaining white solid is dissolved in dichloromethane (150 mL), and the resulting solution transferred into a separation funnel (250 mL) and washed with a saturated solution of NaBF4 (4 × 40 mL) (Note 1). The aqueous phase is extracted with dichloromethane (50 mL), and the combined organic layers are dried with MgSO4 (6 g). The solid drying agent is filtered off and washed with an additional 50 mL of dichloromethane, and the combined filtrates are concentrated on a rotary evaporator (10 mmHg, bath temperature ca. 40 °C) to afford 1,1,3,3-tetrakis(dimethyl-amino)propenium tetrafluoroborate as a beige solid (10.5 g, 77%) (Note 7).
C. Tetrakis(dimethylamino)allene. A 100-mL, three-necked, round-bottomed, flame-dried flask equipped with a teflon-coated stir bar (3 cm), a glass stopper, a tubing adapter connected to an ammonia tank, and a cold finger connected to a gas outlet (Note 8) is charged under argon with 1,1,3,3-tetrakis(dimethylamino)propenium tetrafluoroborate (8.20 g, 27.3 mol, 1.0 equiv) and sodium amide (1.36 g, 35 mmol, 1.2 equiv) (Note 1). The cold finger is filled with a mixture of dry ice/acetone and the flask is immersed into a dry ice/acetone cooling bath (−78 °C) before dry ammonia (ca. 40 mL) (Note 9) is condensed into the flask. The cooling bath is removed, causing the ammonia to reflux back from the cold finger kept at −78 °C into the reaction mixture. After 1 hour, the cooling of the condenser is discontinued such that the ammonia gently evaporates while the reaction mixture reaches room temperature (Note 10). Once the excess ammonia has evaporated, the reflux condenser is quickly replaced by a small distillation head (Note 11) and the residue is distilled in vacuum (bp 41-46 °C/10-3 mmHg; bp 64-68 °C, 0.4 mmHg) (Note 12) to afford tetrakis(dimethylamino)allene as a colorless oil that can be stored at −20 °C under argon for months without noticeable decomposition (4.98 g, 85%) (Notes 13 and 14).
2. Notes
1.
Dichloromethylene-dimethyliminium chloride (technical grade, ≥ 95% Acros),
NaBF4 (98%) (Aldrich Chemical Company, Inc.), and
dimethylamine (≥ 99%, Fluka) were used as received.
Dimethylacetamide (≥ 99%, Aldrich Chemical Company, Inc.) was dried over BaO prior to use and stored over 4 Å molecular sieve; KFC-Titration showed 14 ppm of water.
NaNH2 (technical grade, Aldrich Chemical Company, Inc., ≥ 95% Acros) was used as received and transferred under Ar.
2.
The CH
2Cl
2 was distilled over CaH
2 and transferred under argon.
3.
The product salt is ca. 90% pure and contains impurities mainly derived from the starting material that is technical grade. It is prone to hydrolysis when kept in air and must be stored under argon. Under argon, the compound stays stable for more than a week (if exposed to air, the solid decomposes during ca. 4 h). Characteristic physicochemical properties:
1H NMR
pdf (300 MHz, CDCl
3) δ: 3.55 (s, 12 H), 5.76 (s, 1 H);
13C NMR
pdf (75 MHz, CDCl
3) δ: 44.8, 89.6, 158.8; IR (neat) 2944, 1626, 1559, 1429, 1304, 1204, 1127, 764 cm
-1; HRMS
m/z calcd for C
7H
13N
2Cl
2 195.0456, found 195.0449. An additional broad singlet at 12.07 ppm in the
1H NMR might be due to the technical grade starting material or compounds derived from partial hydrolysis of the product; it did not appear to have an effect on the next reaction.
4.
The Checkers used Teflon tape to mark the line up to which the Schlenk tube was filled. To enhance the condensation of dimethylamine, 80% of the length of the tube was placed inside the cooling bath.
5.
The Checkers found it useful to condense 26 mL of dimethylamine to facilitate stirring at the beginning of the reaction. On small scale (half-scale) this was not necessary.
6.
Excess dimethylamine was destroyed by bubbling the gas stream through dilute
sulfuric acid (250 mL, ca. 0.5 M,).
7.
The product is ≥ 95% pure (NMR) and has the following physicochemical properties:
1H NMR
pdf (400 MHz, CDCl
3) δ: 2.94 (s, 24 H), 3.56 (s, 1 H);
13C NMR
pdf (100 MHz, CDCl
3) δ: 40.9, 72.4, 169.2; IR (neat) 2950, 1526, 1395, 1028, 764, 688 cm
−1; HRMS
m/z calcd for C
11H
25N
4 213.2079, found 213.2078.
8.
The following set-up was used:
9.
Ammonia (anhydrous) was used without further purification.
10.
The evaporation of the ammonia took approximately 2 h after cooling was discontinued
11.
A distillation head with a thermometer and a water-jacketed condenser arm (ca. 5 cm length) was used.
12.
No fractions were taken during the distillation, which was continued until a very thick salt slurry had formed. If distillation was continued beyond that point and the slurry was distilled to dryness, yellow byproducts were also collected in the distillate, and the resulting thick yellow oily distillate was subjected to a second distillation without decreasing the yield. Because the residual slurry may contain traces of unreacted NaNH
2, it was decomposed upon suspension in
hexanes (100 mL) and careful addition of
isopropanol (30 mL), followed by the addition of
water (50 mL).
13.
The product is ≥ 95% pure (NMR) and has the following physicochemical properties:
1H NMR
pdf (300 MHz, C
6D
6) δ: 2.62 (s, 24 H);
13C NMR
pdf (75 MHz, C
6D
6) δ: 42.1, 142.8, 157.0; HRMS
m/z calcd for C
11H
25N
4 (M+H)
+ 213.2079, found 213.2077. The product fumes when exposed to air and must be kept under argon. Because of this sensitivity, an accurate elemental analysis could not be obtained, and the checkers did not measure an IR.
14.
The submitters obtained 3.77-4.52 g (65-78%). IR (neat) 2864, 1894, 1613, 1544, 1507, 1424, 1370, 1326, 1149, 1114, 1021, 924, 762, 681 cm
-1.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Attachment of electron releasing substituents to the termini of an allene induces a significant polarization of the π-bonds towards the central carbon atom.
3 The substantial contribution of the resulting dipolar mesomeric extremes (for example,
1' and
1'') is clearly expressed in the peculiar physical and chemical properties of such compounds.
Specifically, their central C-atoms resonate at unusually high field (compare δ
C = 141.8 ppm in
1 compared with δ
C = 211.4 ppm in the parent allene H
2C=C=CH
2). Even more striking is the fact that tetra-amino(alkoxy)allenes either (strongly) deviate from linearity or can be bent to a very significant degree at almost no energetic cost.
3,4 This propensity allowed such units to be incorporated even into a five-membered (!) ring system by virtue of the fact that the aromatic resonance form
3' holds an important share of the ground state structure.
5Figure 1 Comparison of Allene Ground States
The particular bonding situation of tetraaminoallenes also dominates their chemical behavior. Specifically, compound
1 forms stable dipolar adducts with CO
2 (
4), CS
2 or SO
2,
6 and reacts with metal templates such as [(Ph
3P)Au]
+ to give the structurally unusual η
1-adduct
5 rather than the η
2-bound π-complex to be expected for a normal allene ligand.
7 Furthermore, it has been shown that related tetraaminoallenes can be protonated twice at the central C-atom.
8 These experimental findings are in good accord with computational studies which suggest that lateral donor substituents impart considerable "carbodicarbene" character onto an allene motif. The resonance extreme of a "carbodicarbene" can be described as consisting of a formally zerovalent carbon atom – featuring two orthogonal electron pairs – which is stabilized by coordination to two flanking diaminocarbene units.
3,9Scheme 1
Figure 2 Stabilization of "Carbodicarbene"
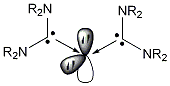
The preparation of tetrakis(dimethylamino)allene as the parent compound of this series described herein is a safe and convenient modification of the original method described by Viehe and coworkers.
6,10 The essential differences are the use of (i) tetrafluoroborate salts instead of perchlorates,
11 and (ii) NaNH
2 in liquid ammonia
12 rather than
n-BuLi in THF as the base for the final deprotonation reaction. The BF
4− counterion ensures that the tetraamino-substituted allyl cation produced in step B is sparingly water soluble and therefore can be easily separated from the dimethyl ammonium chloride byproduct by an aqueous work up. Moreover, the purification of the final product is more convenient when NaNH
2 in liquid ammonia is used,
12 as this combination leaves no organic solvent behind and hence avoids a cumbersome fractionation. Since the generated NaBF
4 salt also does not need to be separated, no further manipulation of the air sensitive tetraaminoallene
1 is necessary; simple distillation of the crude mixture provides the desired compound in analytically pure form in good yield. In contrast, a distillation in the presence of perchlorate salts poses a serious hazard,
11 in particular when performed on a multigram scale. Because the procedure described herein also applies to the preparation of a host of other donor substituted allenes and related cumulated ylides, it is expected to spur the systematic exploration of the fascinating chemical and physical properties of such electron rich compounds, an area of research which was largely dormant until very recently.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Dichloromethylen-dimethyliminium chloride; (33842-02-3)
Dimethyl acetamide; (127-19-5)
1,3-Dichloro-1,3-bis(dimethylamino)propenium chloride; (34057-61-9)
Dimethylamine; (124-40-3)
1,1,3,3-Tetrakis(dimethylamino)propenium tetrafluoroborate; (125254-01-5)
Sodium amide; (7782-92-5)
Tetrakis(dimethylamino)allene; (42928-64-3)
 |
Alois Fürstner was born in Bruck/Mur, Austria, in 1962. He obtained his training and PhD degree from the Technical University of Graz, Austria, working on carbohydrates under the supervision of Hans Weidmann. After postdoctoral studies with the late Prof. W. Oppolzer in Geneva, he finished his Habilitation in Graz before joining the Max-Planck-Institut für Kohlenforschung, Mülheim/Ruhr, Germany, in 1993, where he is presently the Managing Director. His research interests focus on homogeneous catalysis, metathesis, and organometallic chemistry as applied to natural product synthesis. |
 |
Manuel Alcarazo was born in 1978 in Alcalá de Guadaira, Spain. His training as a chemist started at the University of Sevilla where he was graduated in 2000. He obtained his PhD in 2005 at the Instituto de Investigaciones Químicas (CSIC-USe) under the supervision of Prof. Rosario Fernández and Dr. José M. Lassaletta. There he worked on the synthesis and applications of N-heterocyclic carbenes derived from hydrazines. After that he joined Prof. A. Fürstner's research group as a postdoctoral associate at the Max-Plank-Institut für Kohlenforschung where he was involved in various projects ranging from ligand design to natural product total synthesis. He is currently an independent junior scientist at the Max-Planck-Institut für Kohlenforschung working on the design and synthesis of ligands based on low valent p-block elements. |
 |
Helga Krause was born in 1952 in Oberhausen, Germany. She started her education in 1969 as a laboratory assistant at the Max-Planck-Institute for Coal Research. After completing these studies in 1972 she worked in the group of Professor Roland Köster in boron-organic chemistry. In 1994 she joined the group of Professor Alois Fürstner working on various projects in the field of organic and organometallic chemistry. |
 |
Carl Deutsch was born in Dinslaken (Germany) and studied chemistry in Dortmund, where he obtained his Diploma in 2004 and his PhD in 2008 under the supervision of Prof. N. Krause on the CuH-catalyzed synthesis of allenes. Parts of his research, for which he received a DSM Science & Technology Award in 2008, were carried out with Prof. B. H. Lipshutz (Santa Barbara, CA, U.S.A.) and, as JSPS fellow with Prof. M. Murakami (Kyoto, Japan). He is currently a postdoctoral researcher in the group Prof. P. Wipf, Pittsburgh, U.S.A., sponsored by the DFG, and works in the area of bioactive natural product synthesis. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved