Org. Synth. 2025, 102, 442-464
DOI: 10.15227/orgsyn.102.0442
Synthesis of Abietatriene via Nitrogen Deletion
Submitted by Julia L. Driscoll, Carys E. Obertone, Liao Chen, and Mark D. Levin*
1Checked by Yu Chen and Pauline Chiu
1. Procedure (Notes 1, 2)
A. Leelamine acetate (2). In air, 90% (+)-dehydroabietylamine (leelamine, 1) (13.3 g, 42.5 mmol, 1.0 equiv) (Note 3) is added to a 250-mL Erlenmeyer flask, followed by toluene (120 mL) (Note 4), to form a deep yellow solution (Figure 1A). A cylindrical stir bar (4 cm) is added and the solution is heated to an internal temperature of 64 °C (Note 5) while stirring (Note 6). Glacial acetic acid (4.4 mL, 77 mmol, 1.8 equiv) (Notes 7 and 8) in toluene (10 mL) is added to a 30-mL separatory funnel, used as an additional funnel, positioned above the flask (Figure 1B), and this solution is added dropwise to the reaction over 4 min (Notes 9,10,11). Throughout the addition, the solution remains transparent and yellow (Figure 1C).
Figure 1. A. Leelamine dissolved in toluene; B. reaction setup; C. reaction after addition of acetic acid (Photos by the checkers)
After addition is complete, the solution is slowly cooled to 35 °C (Note 12), and a slurry begins to form. This temperature is held for 30 min to achieve slow crystallization, during which a thick slurry formed (Note 13) (Figure 2A). Then the heating is switched off, and the solution is left in the oil bath to cool slowly. Once an internal temperature below 30 °C is achieved (Note 14), the flask is transferred to an ice water bath to cool further for 15 min (Note 15) (Figure 2B).
Figure 2. A. initial slurry formation; B. cooling in ice water bath (Photos by the checkers)
The slurry is filtered under house vacuum (~75 mm Hg) using a 150-mL glass fritted Büchner funnel (Note 16) attached to a 250 mL side-arm filter flask using a rubber filter adaptor (Note 17) (Figures 3A-C). Once the top of the cake shows no visible liquid (Figure 3D), it is rinsed with cold toluene (40 mL x 2) (Note 18) and filtered to afford a white solid (Figures 3E-F).
Figure 3. First filtration: A. vacuum filtration setup; B. filtered slurry and mother liquor; C. mother liquor with material remaining in reaction flask; D. filter cake immediately before washing; E. and F. filter cake after washing (Photos by the authors)
The solids are transferred to a 125-mL Erlenmeyer flask (Figure 4A). Toluene (75 mL) and a cylindrical stir bar (4 cm) are added, and the resulting white suspension (Figure 4B) is heated to 65 °C to form a clear, colorless solution (Note 5) (Figure 4C). Once the solids are fully dissolved, the solution is slowly cooled to 49 °C (Note 19), and a slurry began to form (Note 20). This temperature is held for 30 min while the slurry thickened (Figure 4D) before allowing the bath and solution to cool at room temperature (Notes 14, 21). Once the internal temperature reaches 30 °C, the flask is placed in an ice water bath for 15 min (Note 15) (Figures 4E-F).
Figure 4. Recrystallization: A. filtered solids in flask; B. suspension in toluene; C. solution at 65 °C; D. slurry progression at 49 °C; E. cooling in ice water bath; F. final slurry (5 °C) (Photos by the authors)
The slurry is filtered under house vacuum (~75 mm Hg) using a 150-mL glass fritted Büchner funnel (Note 16) attached to a 250 mL side-arm filter flask using a rubber filter adaptor (Note 17) (Figure 5A). Once no liquid is visible on top of the cake (Figure 5B), it is rinsed with cold toluene (40 mL x 2) followed by cold pentane (30 mL) (Note 22) to afford a white solid (Figures 5C-D). When the filter is no longer cold to the touch, the cake is broken up by scraping with a plastic spatula to form chalky white solids (Figure 5E). Once dry, the solids are transferred to a 250 mL round-bottom flask and dried under vacuum overnight (Note 23) to afford 2 as a chalky white solid (7.05 g, 20 mmol, 48%) (Notes 24,25,26) (Figure 5F).
Figure 5. Second filtration: A. slurry in funnel; B. filter cake immediately before rinses; C. and D. dried filter cake; E. solids broken up; F. dried solids (Photos from the authors)
B. Abietatriene (3). A Teflon-coated, egg-shaped magnetic stir bar (3 × 1.5 cm) was placed inside a three-necked, 500-mL (TS 14/20, 24/40, 14/20) round-bottomed flask. The assembly was oven-dried at 120 °C overnight (14 h). It is removed from the oven and fitted with a thermometer adapter and a thermometer on one side-neck. The center neck was fitted (a TS 14/20 to 24/40 joint adapter was used) with an oven-dried, 100-mL pressure-equalizing addition funnel (TS 14/20, 14/20) with a Teflon stopcock. All the joints were secured using Keck clips. The addition funnel, as well as the third neck of the flask, are sealed with rubber septa. Each septum is pierced with a needle connected to an argon line, and another needle connected to a gas outlet terminating in an oil bubbler. The entire reaction assembly is allowed to cool to room temperature (~ 22 °C) under a flow of dry argon (Note 27). (Figure 6A).

Figure 6. A. Reaction setup; B. Compound 2 in dry MeCN; C. N-(Benzyloxy)-N-(pivaloyloxy)-4-(trifluoromethyl)benzamide is transferred to the addition funnel via cannula (Photos provided by the checkers)
With the argon gas inlets intact, leelamine acetate 2 (5.15 g, 15 mmol, 1.0 equiv) is added to the flask under a flow of argon by temporarily removing the rubber septum of the third neck. The septum is replaced. Dry, degassed acetonitrile (100 mL) (Note 28) is added by syringe. The flask is placed in an ice-water bath with stirring (Note 29) until a white suspension equilibrated to 3 °C was obtained (Figure 6B)
N-(Benzyloxy)-N-(pivaloyloxy)-4-(trifluoromethyl)benzamide 4 (6.49 g, 16.4 mmol, 1.1 equiv) (Note 30) is placed in an oven-dried 100 mL heart-shaped flask (TS 14/20), and the flask is sealed with a rubber septum (Figure 6C). The flask is pierced with a needle connected to an argon line and another needle connected to a gas outlet terminating in an oil bubbler. The sample was purged with argon for 10 min. Then dry, degassed acetonitrile (40 mL) (Note 28) is added to the flask, and once the solid is fully dissolved, the solution is transferred to the closed addition funnel via cannula (Note 31) (Figure 6C). The heart-shaped flask is then rinsed with acetonitrile (5 mL), and the rinse is transferred via cannula to the addition funnel.
The solution in the addition funnel is added to the white suspension dropwise at a rate of 2 drops per second (ca. 20 min). Steady gas evolution is observed during the addition, and the rate of addition is controlled to minimize effervescence and to ensure that the internal temperature does not increase above 5 °C. After the addition is complete, 5 mL of dry acetonitrile is added slowly by syringe to rinse down the sides of the addition funnel. The rinse is added to the reaction, and the reaction is left to stir at 3 °C for 3 h, until most of the solids are dissolved and the solution has a slightly translucent appearance. At this point, the ice-water bath is removed, allowing the reaction to reach room temperature (22 °C) (Note 32).
After 1 h of stirring at room temperature, the reaction is nearly transparent (Figure 7). Triethylamine (4.2 mL, 30 mmol, 2 equiv) (Notes 33-34) is added by syringe over 1 min, during which a slight exotherm was observed (25 °C). The reaction is stirred for another 30 min until full consumption of 2 was observed by TLC (Note 35).
Figure 7. Reaction mixture after stirring for 1 h at room temperature (Photo provided by the checkers)
After the reaction is determined to be complete, the thermometer and stir bar are removed, rinsing each with ~ 2 mL of hexanes into the reaction flask. The contents of the flask are transferred to a 500-mL separatory funnel. The reaction flask is rinsed thrice with hexanes (100 mL divided into 3 portions) (Note 36), and these are also transferred to the funnel. The funnel is vigorously shaken.
After separating the hexanes and acetonitrile layers, the acetonitrile layer is extracted with more hexanes (2 x 100 mL). The combined hexanes layers are then washed sequentially with deionized water (100 mL) and brine (50 mL), followed by vacuum filtration through anhydrous sodium sulfate (20 g) (Note 37) in a 150 mL fritted Büchner funnel, into a 1 L round-bottom flask. The funnel is rinsed with additional hexanes (10 mL x 3) which were added to the round-bottom flask. The hexane is then removed via rotary evaporation (Note 38) to afford a pale-yellow oil (Figure 8).
Figure 8. Crude product as an oil (Photo provided by the authors)
The crude residue is diluted with hexanes (5 mL) (Note 36) and loaded onto a column (2.25 x 8 in) packed with 235 g silica gel (Note 39) and equilibrated with hexanes, rinsing the flask with additional hexanes (up to 3 mL) to transfer all of the material. The column is then eluted with 1 L of hexanes, and 50 mL fractions were collected. Based on TLC analysis (Notes 40,41,42), fractions 11-17 are combined and concentrated by rotary evaporation (Note 38), then placed under high-vacuum (Note 23) for 6 h to afford abietatriene as a colorless oil (3.24 g, 80% yield, Notes 43,44,45) (Figure 9A). Upon storage at -22 °C overnight, the oil crystallized to a white solid (Figures 9B-C).
Figure 9. Purified product: A. oil; B and C. crystallized (Photos by the authors)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of “Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also “Identifying and Evaluating Hazards in Research Laboratories” (American Chemical Society, 2015) which is available via the associated website “Hazard Assessment in Research Laboratories” at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
leelamine,
acetic acid,
N-(benzyloxy)-N-(pivaloyloxy)-4-(trifluoromethyl)benzamide,
triethylamine,
toluene,
pentane,
acetonitrile, hexanes, and brine. Special caution should be taken with the storage and handling of
4, as a DSC has shown that it decomposes when heated.
2 Additionally, Ames testing has demonstrated that some anomeric amide derivatives are mutagenic.
3,4,5,6,7 The mutagenicity of
4 was recently examined using Ames II testing. Compound
4 exhibits mutagenicity lower than other members of the anomeric amide reagent class and on par with other commonly handled laboratory reagents such as benzyl chloride.
8 Nonetheless, appropriate ventilation and personal protective equipment should be employed when handling
4. Additionally, due to the generation of
N2 gas in step b of this procedure, careful attention at the start of the addition of
4 to the reaction and while warming the is essential to prevent rapid gas generation.
2. Part A of the procedure was adapted from the procedure of Lee C. Cheney.
93.
(+)-Dehydroabietylamine (60%) was purchased from Sigma Aldrich and used as received. The checkers purchased
(+)-dehydroabietylamine (90%) from Shanghai Aladdin Bio-Chem Technology Co., Ltd., which was used as received. The purity of
1 received was found to be 91% by the checkers by qNMR using
1,3,5-trimethoxy benzene (99.9%, Sigma Aldrich) as the internal standard.
4.
Toluene (certified ACS, 99.5%) was purchased from Fisher and used as received. The checkers purchased
toluene (GR grade) from Duksan Pure Chemicals Co. Ltd, which was used as received.
5. The authors set the bath temperature to 66 °C to achieve this internal temperature.
6. A stirring rate of 180 rpm was used.
7. Glacial
acetic acid (HPLC grade, Supelco) was purchased from Millipore Sigma and used as received. The checkers purchased glacial
acetic acid (>99.9%, HPLC grade) from Shanghai Macklin Biochemical Co., Ltd., which was used as received.
8. Technical grade
leelamine (60%) which was purchased from Sigma Aldrich by the authors contains mostly high molecular weight primary amines.
10 Assuming these amines have a molecular weight similar to
leelamine, 1.75 equiv
acetic acid relative to
leelamine equates to 1.05 equiv relative to the total amount of primary amine compounds.
9. The solution was added at a rate of 2-3 drops/second.
10. After the completion of the
acetic acid addition the pH of the solution was checked using a universal pH indicator strip wetted with DI water to ensure that the solution was acidic. If the solution is not acidic after this addition,
acetic acid should be added dropwise until the pH is below 7.
11. The solution reached maximum internal temperature of 67 °C during acid addition.
12. The solution was cooled by lowering the temperature of the oil bath to a setting of 37 °C. An internal temperature of 37 °C was reached 45 min after the set temperature was changed.
13. The stirring rate was increased to 550 rpm (until top of slurry is incorporated).
14. The solution reached an internal temperature of 30 °C after 15 min.
15. An internal temperature of 5 °C was reached.
16. The funnel used was a Synthware 150-mL Buchner funnel with a fine porosity glass frit.
17. After the initial filtration, the mother liquor was used to rinse the original flask and passed through the filter to transfer as much of the solids as possible (Figure 3C).
18.
Toluene was cooled in an ice water bath for at least 10 min prior to use.
19. The solution was cooled by lowering the temperature of the oil bath to a setting of 50 °C. An internal temperature of 49 °C was reached 45 min after the set temperature was changed.
20. The stirring rate was increased to 400 rpm (until top of slurry is incorporated).
21. The stirring rate was increased to 500 rpm.
22.
Pentane (certified, 98%) was purchased from Fisher and used as received. The checkers purchased
pentane (GR grade) from Duksan Pure Chemicals Co. Ltd., which was used as received.
23. The product was dried at room temperature (22 °C) under high vacuum (<1 mm Hg) for 4 h.
24. The purity of
2 was determined to be 99% by qNMR using
1,3,5-trimethoxybenzene (99.8%, Acros, used as received) as an internal standard. The purity of
2 was determined to be 99% by the checkers by qNMR
pdf using
1,3,5-trimethoxybenzene (99.9%, Sigma Aldrich, used as received) as an internal standard.
25. Product
2 is characterized as follows:
1H NMR
pdf (400 MHz, CD
3OD) δ 7.16 (d,
J = 8.2 Hz, 1H), 6.95 (d,
J = 8.1 Hz, 1H), 6.87 (s, 1H), 3.00 - 2.67 (m, 5H), 2.37 (d,
J = 12.8 Hz, 1H), 1.89 (s, 3H), 1.87-1.73 (m, 4H), 1.56 (d,
J = 12.9 Hz, 1H), 1.49 - 1.31 (m, 3H), 1.23 (s, 3H), 1.19 (d,
J = 6.9 Hz, 6H), 1.06 (s, 3H) ppm;
13C NMR
pdf (151 MHz, CD
3OD) δ 180.1, 148.0, 147.0, 135.5, 127.7, 125.1, 124.9, 51.9, 47.3, 39.3, 38.6, 37.0, 36.4, 34.8, 30.6, 25.6, 24.50, 24.46, 24.1, 20.0, 19.4, 18.3 ppm; mp: 146.7-146.9 °C; IR (diamond ATR, neat) 2927, 1528, 1393, 1333, 1010, 883, 822, 649, 470 cm
-1; HRMS (ESI)
m/z calculated for [M+H]
+ (C
20H
32N
+): 286.2607, found: 286.2605.
26. A second, half-scale reaction performed by the checkers provided 3.82 g (50% yield) of compound
2 of 99% purity as determined by qNMR using trimethoxybenzene as an internal standard.
27. The authors cooled the reaction apparatus under vacuum and refilled with
nitrogen gas. After addition of
2, the assembly was subjected to three cycles of evacuation and refilling with nitrogen before beginning the reaction. The checkers assembled the apparatus under an active pressure of
argon.
28.
Acetonitrile (HPLC grade, Fisher) was degassed under
argon and dried by passing through a PPT Solvent Purification System. The checkers purchased
acetonitrile (super dry, water < 10 ppm) from J&K Scientific Ltd., which was sparged for 15 min with
argon before use.
29. A stirring rate of 200 rpm was used.
30. The authors used
N-(benzyloxy)-N-(pivaloyloxy)-4-(trifluoromethyl) benzamide 4 (90% purity), which was prepared following an
Organic Syntheses procedure.
11 The checkers used freshly prepared
4 of 97% purity as assessed by qNMR.
31. For the cannula transfer, the gas outlet is removed from the heart-shaped flask, and the gas inlet is removed from the top of the addition funnel to establish the pressure differential for transfer.
32. The reaction should be visually monitored for 3 min after removal of the ice water bath. Mild gas evolution is typical during this stage, but if vigorous gas evolution is observed, the reaction should be placed in the ice water bath to stir for another 30 min.
33.
Triethylamine (reagent grade, Fisher) was degassed under
argon and dried by passing through a PPT Solvent Purification System. The checkers purchased
triethylamine (> 99.0%) from TCI Co. Ltd., which was used as received.
34. Two equivalents of
triethylamine are used to neutralize the
acetic acid released by
2, and the pivalic acid generated from
N-(Benzyloxy)-N-(pivaloyloxy)-4-(trifluoromethyl)benzamide in the reaction.
35. Reaction progress was monitored by TLC using 20% v/v ethyl acetate in hexanes. The plate should be visualized using UV light and ninhydrin stain to detect the disappearance of
dehydroabietylamine (baseline spot). (Figure 10)
Figure 10. TLC of completed reaction (20% EA/hexanes) A. under UV (UV-active spots circled with pencil); B. ninhydrin stain; C. back of stained plate (inverted for consistency) (Photos provided by the authors)
36. Hexanes (certified ACS, 98.5%) was purchased from Fisher and used as received. The checkers purchased hexanes (GR grade) from Duksan Pure Chemicals Co. Ltd, which was used as received.
37.
Sodium sulfate, anhydrous (99.5%) was purchased from Oakwood Chemical and used as received. The checkers purchased anhydrous
sodium sulfate (AR grade) from Dieckmann (Hong Kong) Chemical Industry Co. Ltd., which was used as received.
38. A rotary evaporator was used with a bath temperature of 30 °C from 350 mm Hg to 200 mm Hg.
39. SiliaFlash F60 (particle size 40-63 μm) was purchased from SiliCycle and used as received. The checkers purchased silica gel (0.040 - 0.063 mm, 230 - 400 mesh) from Merck Millipore, which was used as received.
40. Column fractions were analyzed by TLC using hexanes. The plate should be visualized by UV and by
p-anisaldehyde (PAA) stain to detect closely eluting UV-inactive byproducts.
41. Fractions 11-17 were collected to obtain the desired product
3. Fractions 10, 18, and 19 were excluded to ensure high product purity due to the detection of byproducts. (Figure 11)
Figure 11. TLC of column fractions A. under UV; B. with PAA stain (Photos provided by the authors)
42. In some trials it was found that when product concentration was high in mixed fractions, the inclusion of early mixed fractions (Figure 12, fractions 14 and 15) was found to have minimal effect on the purity with improved yield. (Fractions 16-22: 55% yield, 100% purity; Fractions 14-22: 77% yield, 99.5% purity). (Figure 12)
Figure 12. TLC of column fractions from replicate reaction with PAA stain (Photos provided by the authors)
43. The purity of
3 was determined to be 99 wt% by qNMR
pdf using
1,3,5-trimethoxybenzene (99.8%, Acros, used as received) as an internal standard. The checkers determined the purity of
3 to be 98% by qNMR
pdf using
1,3,5-trimethoxy benzene (99.9%, Sigma Aldrich, used as received) as an internal standard.
44. Product
3 is characterized as follows:
1H NMR
pdf (400 MHz, CDCl
3) δ 7.18 (d,
J = 8.1 Hz, 1H), 6.99 (dd,
J = 8.1, 2.1 Hz, 1H), 6.89 (d,
J = 2.2 Hz, 1H), 2.98 - 2.76 (m, 3H), 2.28 (dd,
J = 12.8, 3.8 Hz, 1H), 1.91 - 1.85 (m, 1H), 1.82 - 1.56 (m, 3H), 1.56 - 1.31 (m, 3H), 1.25-1.21 (m, 7H), 1.19 (s, 3H), 0.94 (d,
J = 6.4 Hz, 6H) ppm;
13C NMR
pdf (151 MHz, CDCl
3) δ 147.8, 145.5, 135.1, 127.0, 124.4, 123.9, 50.6, 41.9, 39.0, 37.7, 33.6, 33.5, 30.7, 25.0, 24.2, 24.1, 21.8, 19.5, 19.3 ppm; mp: 46.7-46.9 °C. IR (diamond ATR, neat) 2930, 1497, 1456, 1373, 824, 630, 483 cm
-1; HRMS (ESI)
m/z calculated for [M]
- (C
20H
30-): 270.2298, found: 270.2297.
45. A second, half-scale reaction performed by the checkers provided 1.61 g (82% yield) of compound
3 of 98% purity as determined by qNMR using trimethoxybenzene as an internal standard.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
This reaction demonstrates the use of anomeric amide reagent
4 to enable the direct deamination of leelamine (dehydroabietylamine) (
1) to access abietatriene in a single step in good yield. Abietatriene is a naturally occurring diterpene that has been synthesized as an intermediate en route to a variety of terpenoid natural products and their synthetic analogs (Figure 13).
12,13,14,15,16,17,18,19,20,21Figure 13. Compounds synthesized from abietatriene
Due to its limited commercial availability, abietatriene is typically synthesized from leelamine (
1) or dehydroabietic acid (
5). Prior to the development of the above reaction, the most popular route to achieve this has involved transforming the amine or carboxylic acid respectively into an aldehyde (
6), followed by a Wolff-Kishner type reduction (Scheme 1a).
12,13,14,15,22 From leelamine (
1), the aldehyde intermediate can be accessed using pyridinium salt
7,
14,15 based on a biomimetic transformation developed by Rapoport.
23 From dehydroabietic acid, aldehyde
6 has been synthesized by conversion to Weinreb amide
8 (via acyl chloride
9) followed by hydride reduction.
13 Alternatively, the carboxylic acid can be fully reduced to an alcohol (
10) followed by pyridinium chlorochromate oxidation.
12,22 Alcohol
10 can also be tosylated (
11) then reduced using sodium iodide and zinc dust.
16,17 A more direct method employing hydroxylamine-
O-sulfonic acid (HOSA) for the direct reduction of the amine has been used to achieve moderate yield on a phenolic derivative (
12 and
13) (Scheme 1b).
14,19 Other synthetic strategies from leelamine have included reduction of the tosylated amine (
14) using base followed by ethereal chloramine
24 and formylation to
15 followed by dehydration to generate an isonitrile (
16), which can be reduced using potassium (Scheme 1b).
18 In addition to these reduction strategies,
de novo construction of the B ring has also been demonstrated from various combinations of cyclohexene-based and aromatic fragments (Scheme 1c).
20,21,25
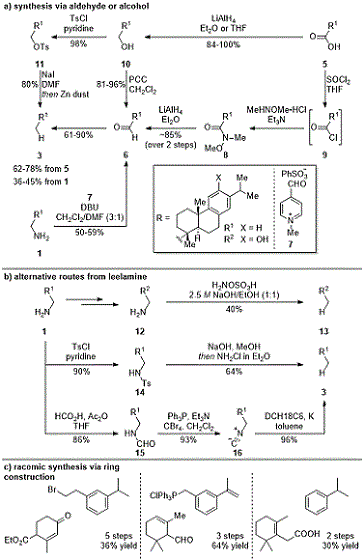
Scheme 1. A. synthetic routes to 3 proceeding through an aldehyde or alcohol intermediate; B. alternative synthetic routes to 3 from leelamine; C. fragment pairs for the synthesis of 3 via ring construction strategies
This method is applicable to the direct deamination of many other primary amines as described in the discussion for the synthesis of
411 Deviations from the published standard conditions for this transformation
2 for the large scale synthesis of
3 include the use of the acetate salt (
2) rather than the free amine (
1), lowering of the addition temperature to 0 °C, the addition of triethylamine, and direct extraction into hexanes from the reaction solvent (Table 1). The reaction of
1 with primary amines was observed to be very rapid on small scale, with visible gas evolution. The use of the acetate salt and lower addition temperature enable better control over the rate at which nitrogen gas is generated throughout the reaction. Additionally, the conversion of
1 to an acid salt is necessary for its purification from the commercially supplied technical grade material, so the direct deamination of the acetate salt increases the efficiency of this route by eliminating the need to free-base the amine. Due to the reduced solubility of the acetate salt in acetonitrile and the acidification of the solution over the course of the reaction from the generation of 1 equivalent of pivalic acid, triethylamine was added to ensure complete consumption of the amine and removal of acidic byproducts during extraction. Product isolation is further simplified by extracting directly into hexanes from the crude reaction mixture, which was found to efficiently extract the product and remove polar byproducts while eliminating the need to evaporate the acetonitrile.
Table 1. published general conditions for primary amine deletion2 vs. these conditions
Appendix
Chemical Abstracts Nomenclature (Registry Number)
(+)-Dehydroabietylamine (leelamine); (1)(1446-61-3)
Acetic acid; (64-19-7)
N-(Benzyloxy)-N-(pivaloyloxy)-4-(trifluoromethyl)-benzamide; (2648454-31-1)
Triethylamine: N,N-diethyl-ethanamine; (121-44-8)

|
Julia L. Driscoll is originally from Pittsburgh, Pennsylvania. She received a B.S. in Chemistry from the University of Pittsburgh where she worked on the synthesis of complex bioactive molecules with Kazunori Koide. Julia is currently a graduate student in the Levin Group focusing on mechanistic elucidation and reagent development for nitrogen-deletion reactions. |

|
Carys E. Obertone is from Atlanta, Georgia. She attended Emory University, where she obtained a B.S. in Chemistry while studying fibrinogen and clotting characterization in COVID-19 patients under Dr. Cheryl Maier. Carys is currently pursuing her Ph.D. in Chemistry in the Levin Group at the University of Chicago, researching deaminative strategies towards the alteration of biological molecules. |

|
Liao Chen is from Shanghai, China. He currently attends the University of Chicago as an undergraduate student and is expected to receive a Bachelor of Science degree in Chemistry in 2026. He joined the Levin group in January 2023 when he began his research interest in various nitrogen insertion, deletion, and transmutation methods. |

|
Mark D. Levin, originally from Cleveland, Ohio, is a Professor of Chemistry at the University of Chicago, where his group develops new organic reactions. Mark obtained a B.S. in chemistry from the University of Rochester studying with Alison J. Frontier, a Ph.D. in chemistry from the University of California, Berkeley studying with F. Dean Toste, and conducted postdoctoral research with Eric N. Jacobsen at Harvard University. |

|
Yu Chen was born in Anhui, Mainland China. She received her B.Sc. in 2013 from Anhui University. In 2020, she completed her Ph.D. at the University of Hong Kong under the supervision of Professor Pauline Chiu. Currently, she is pursuing postdoctoral research at the University of Hong Kong, focusing on organic synthesis and medicinal chemistry. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved