Org. Synth. 2025, 102, 582-600
DOI: 10.15227/orgsyn.102.0582
Palladium-Catalyzed Carbonylative Spirolactonization of Hydroxycyclopropanols
Submitted by Cyrus C. Gudeman, Courtney M. Wiethorn, and Mingji Dai*
1Checked by Yusuke Kanno, Juri Sakata, Hidetoshi Tokuyama
1. Procedure (Note 1)
A. 3,3-Dibenzyldihydrofuran-2(3H)-one (1). A three-necked, 500-mL round-bottomed flask (29/32 joint) is equipped with a 3-cm football shaped Teflon-coated magnetic stir bar. Each joint is fitted with a rubber septum. The flask is attached to an argon/vacuum line. The reaction setup is dried by heating the flask with a hot air gun under vacuum (<1 mm Hg) and allowed to cool to room temperature (Note 2). The flask is then backfilled with argon. The right rubber stopper is replaced with a ground glass adapter (29/32 to 15/25) fitted with a Teflon-coated thermometer adapter (15/25) and an ultra-low temperature thermometer (-100 to 50 °C). An argon inlet needle (18G) is inserted into the left stopper and an outlet needle (18G) attached to a bubbler is inserted into the center septum (Figure 1A). The reaction vessel is then charged with bis(trimethylsilyl)amine (24.0 mL, 18.5 g, 115 mmol, 2.2 equiv) (Note 3). To the flask is added anhydrous THF (100 mL) (Note 4). The reaction flask is then placed in a dry ice and acetone bath while stirring (350 rpm) (Note 5). Once the internal temperature reaches -75 °C, n-butyllithium (2.3 M solution in cyclohexane, 50.0 mL, 115 mmol, 2.2 equiv) (Note 6) is added dropwise over the course of 70 min using a syringe pump, taking care to keep the internal temperature below -70 °C. The solution is stirred at -75 °C (internal temperature) for 40 min. Then γ-butyrolactone (3.97 mL, 4.49 g, 52.1 mmol, 1.00 equiv) (Note 7) is added dropwise over the course of 15 min using a syringe pump, taking care to keep the internal temperature of the reaction below -70 °C. The reaction solution is then stirred at -75 °C (internal temperature) for 35 min. To the reaction is then added benzyl bromide (13.6 mL, 19.6 g, 115 mmol, 2.2 equiv) (Note 8) dropwise over the course of 20 min using a syringe pump, taking care to keep the internal temperature below -70 °C. During the addition, the reaction takes on a faint yellow color. The reaction is stirred at -75 °C (internal temperature) for 45 min, then removed from the cooling bath and allowed to warm to room temperature over the course of 45 min. As the reaction warms to room temperature, the reaction mixture becomes more yellow and turbid. The reaction is stirred at room temperature for 18 h, during which time the turbidity of the solution dissipates (Figure 1B).
The argon inlet and septum are removed, and the reaction is exposed to air. The reaction is then quenched by adding water (80 mL), this causes a minor temperature increase of ~5 °C. The reaction mixture is transferred to a 500-mL separatory funnel, and the water layer is extracted three times with ethyl acetate (150 mL, 50 mL, and 50 mL) (Note 9). The organic layers are combined and washed twice with water (100 mL and 50 mL) and once with brine (100 mL). The organic layer is dried over sodium sulfate (50 g) (Note 10). The sodium sulfate is removed by suction filtration (300 mm Hg) through a funnel fitted with a cotton plug, and the sodium sulfate cake is washed twice with 50 mL of ethyl acetate. The solvent is removed by rotary evaporator (<1 mm Hg, 30 °C). The resultant pale-yellow solid is dissolved in 50 mL ethyl acetate and filtered through a plug of Celite (5 g) (Figure 1C) (Note 11). The plug is washed with ethyl acetate (3 x 15 mL) (Note 9), and the solvent is removed via rotary evaporation (<1 mm Hg, 30 °C). The resultant solid is taken up in 50 mL hot dichloromethane (40 °C) (Note 12). The solution is transferred to a 100-mL Erlenmeyer flask, where the solution is heated to boiling until about 10 mL of the solvent boils away (Figure 1D). The solution is then allowed to cool to room temperature (Figure 1E), then cooled to -25 °C in a freezer overnight.

Figure 1. A. The reaction setup; B. The reaction after being stirred overnight at rt; C. The celite plug; D. The recrystallization setup at 34 °C; E. The crystals forming in the flask at room temperature; F. The recrystallized product (photos D and E provided by authors, photos A, B, C and F provided by checkers)
The resulting white crystals are collected by suction filtration (300 mm Hg) using a Buchner funnel and a 5.5-cm diameter filter paper (No. 1). The crystals are washed twice with 15 mL ice-cold diethyl ether (Note 13). The crystals are transferred to a 20-mL scintillation vial and further dried under vacuum (<1 mm Hg) in desiccator overnight to give the first crop of 3,3-dibenzyldihydrofuran-2(3H)-one (1) (9.99 g) as white crystals. The mother liquor is transferred to a round bottom flask, and the solvent is removed via rotary evaporation (<1 mm Hg, 30 °C). The resultant solid is taken up in 30 mL hot dichloromethane (40 °C). The solution is transferred to a 100-mL Erlenmeyer flask, where the solution is heated to boiling until about 10 mL of the solvent boils away. The solution is cooled to room temperature and then to -25 °C in a freezer overnight. The resulting white crystals are collected via suction filtration (300 mm Hg) using a Buchner funnel and a 5.5-cm diameter filter paper (No. 1). The crystals are washed twice with 15 mL ice-cold diethyl ether (Note 13). The crystals are transferred to a 20-mL scintillation vial and dried under vacuum (<1 mm Hg) overnight to give the second crop of compound 1 (1.32 g) as white crystals. The first and the second crops are combined to give compound 1 (11.3 g, 42.5 mmol, 81%) (Figure 1F) (Note 14).
B. 1-(2-Benzyl-4-hydroxy-1-phenylbutan-2-yl)cyclopropan-1-ol (2). A three-necked, 500-mL round-bottomed flask (three necks 29/32 joint) is equipped with a 3-cm football shaped Teflon-coated magnetic stir bar. Each joint is fitted with a rubber septum. The flask is attached to an argon/vacuum line. The reaction setup is dried by heating with a hot air gun under vacuum (<1 mm Hg) and allowed to cool to room temperature (Note 2). The flask is then backfilled with Ar. The right rubber stopper is replaced with a ground glass adapter (29/32 to 15/25) fitted with a Teflon-coated thermometer adapter (15/25) and an ultra-low temperature thermometer (-100 to 50 °C). The middle neck is fitted with an argon inlet. The left neck is fitted with an argon outlet needle attached to a bubbler (Figure 2A). To the flask is added anhydrous THF (160 mL) (Note 15) through the left neck. Then Ti(Oi-Pr)4 (11.2 mL, 10.7 g, 37.6 mmol, 1 eq) (Note 16) is added using a syringe to the reaction vessel while stirring (350 rpm). Then, to the rapidly stirring solution (500 rpm) is added MeMgBr (3.0 M solution in diethyl ether, 19.0 mL, 1.50 eq) (Note 17) dropwise over the course of 20 min (Figure 2B) using a Catamaran syringe pump (Techno Applications Co., Ltd). The solution turns a light green color, and the internal temperature rises from room temperature to 30 °C (Figure 2C).

Figure 2. A. The reaction setup; B. The addition of the MeMgBr; C. The solution after the addition of MeMgBr (Photos provided by checkers)
The reaction is stirred for an additional 10 min before the reaction is placed in an ice and brine bath (Figure 3A). As the reaction cools, it becomes cloudy. Once the internal temperature reaches -2 °C, 3,3-dibenzyldihydrofuran-2(3H)-one (1) (10.0 g, 37.6 mmol, 1.00 eq) is added in ca. 1.5 g portions over the course of 25 min. The solid is added through the left neck of the flask. To the reaction is added EtMgBr (3.0 M solution in diethyl ether, 22.0 mL, 1.7 equiv) (Note 18) via Catamaran syringe pump, dropwise over the course of 4 h, taking care to keep the internal temperature below 4 °C. During the addition of the EtMgBr, the reaction turns brown, then black, and a sludge forms. The reaction is then stirred rapidly (800 rpm) at 0 °C for 3.5 h until the reaction shows complete consumption of the starting material by TLC (Figure 3B) (Note 19). Then to the reaction flask is added 1M sulfuric acid (140 mL) dropwise over the course of 35 min using a pipet, taking care to keep the internal temperature below 20 °C. Care should be taken during this step as the quench is very exothermic and seems to cause gas evolution.
The first 10 mL of the sulfuric acid solution should be added very slowly, and the rate increased gradually thereafter. The reaction mixture is added to a 500-mL separatory funnel, and the aqueous layer is extracted three times with ethyl acetate (100 mL, 100 mL, and 50 mL) (Note 9). The organic layers are combined and washed with brine (100 mL). The organic layer is dried over sodium sulfate (80 g) (Note 10). The sodium sulfate is removed by suction filtration (300 mm Hg) through a cotton plug, and solvent is removed via rotary evaporation (<1 mm Hg, 30 °C). The resultant pale-yellow oil is loaded onto a chromatography column (39 cm height x 6 cm I.D.) containing silica gel (180 g) (Note 20). The product is eluted with a 25% mixture of ethyl acetate in hexane (Note 21) in 32 fractions, each containing 100 mL of solvent.
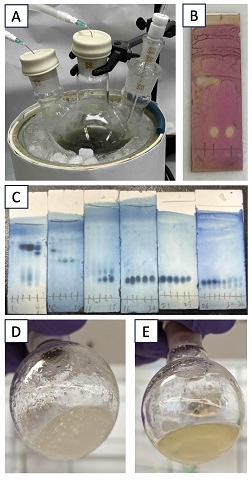
Figure 3. A. The reaction setup in the ice bath (photo provided by checkers); B. The TLC of the reaction. The left spot is the starting material, the middle spot is a co-spot of the starting material and the reaction mixture, the right spot is the reaction mixture; C. The TLC of the column fractions; D. The product collected from fractions 19-25; E. The product collected from fractions 13-18 and 26-32 (Photos D and E provided by authors and photos A, B, and C provided by checkers)
Fractions 13 through 32 contain the desired product. Fractions 13 through 18 and 26 through 32 show other products on TLC (Note 22) (Figure 3C); these fractions are collected separately from fractions 19-25, which contain only the desired product by TLC. The specified fractions are transferred to 500-mL round bottom flasks and the solvent is removed via rotary evaporation (<1 mm Hg, 30 °C) in portions. Care should be taken when removing the solvent, as the product is a highly viscous oil and prone to foaming. The resulting pale-yellow oils are transferred into separate, tared, 50-mL round bottom flasks with diethyl ether (10 mL each). The solvent is again removed via rotary evaporation (<1 mm Hg, 30 °C). The polar solvents, ethyl acetate and diethyl ether, are very difficult to remove from the highly viscous oils. The products are then further dried under vacuum (<1 mm Hg) at 80 °C overnight. Fractions 19-25 yields a colorless oil that contains 3.15 g 1-(2-benzyl-4-hydroxy-1-phenylbutan-2-yl)cyclopropan-1-ol (2) (3.54 g, 89 wt% purity as determined by qNMR (Figure 3D) (Note 23). Fractions 13-18 and 26-32 yields a pale-yellow oil that contains 3.69 g of 2 (4.98 g x 74 wt% purity as determined by qNMR (Figure 3E) (Note 23). Approximately 5 mg of the product is purified further for characterization (Note 24).
C. 9,9-Dibenzyl-1,6-dioxaspiro[4.4]nonan-2-one (3). A single-necked, 1-L round-bottom flask is equipped with an 8 cm Teflon-coated, magnetic stir bar. The mouth of the flask (29/32) is fitted with a septum and placed under vacuum (<1 mm Hg). The flask and stir bar are then heated using a heat air gun to drive any moisture away. The flask is allowed to cool to room temperature under vacuum. The flask is then filled with Ar. To the flask is then added benzene (440 mL) (Note 25). Then 1-(2-benzyl-4-hydroxy-1-phenylbutan-2-yl)cyclopropan-1-ol (2) (3.54 g, 89 wt%, 10.6 mmol, 1.00 equiv) is transferred to the reaction flask using benzene (3 x 20 mL) (Figure 4A) (Note 25). To the solution is added DDQ (4.86 g, 21.2 mmol, 2.00 equiv) (Notes 26 and 27). The solution turns a deep orange color (Figure 4B). The reaction is then placed under vacuum (<1 mm Hg) with stirring (350 rpm) and backfilled with CO (Note 28) from a balloon three times (Figure 4C) (Note 29). The CO atmosphere is maintained with a balloon during the course of the reaction. To the reaction vessel is then added palladium acetate (242 mg, 1.06 mmol, 0.10 eq) in one portion (Figure 4D) (Note 30). The reaction is stirred under CO atmosphere for 5 h. During the course of the reaction, the mixture becomes cloudy (Figure 4E). The progress of the reaction is traced via TLC (Figure 4F) (Note 31).

Figure 4. A. The reaction setup before the addition of DDQ; B. The reaction setup after the addition of DDQ; C. The reaction setup for the evacuation and CO backfill; D. The reaction immediately after the addition of Pd(OAc)2; E. The reaction after 5 h of stirring; F) The TLC of the reaction. The left spot is the starting material, the middle spot is a co-spot of the starting material and the reaction mixture, the right spot is the reaction mixture. (Photos A-E provided by authors, photo F provided by checkers) provided by checkers)
To the reaction is added triethylamine (23 mL) using a syringe (Note 32). The reaction mixture immediately turns black in color. The reaction mixture is stirred for 45 min and then filtered through a two layered plug of Celite (6 g) and silica gel (15 g) (Figure 5A) (Notes 11 and 20). The plug is washed with ethyl acetate(7 x 50 mL) (Note 9). The solvent is then removed via rotary evaporation (<1 mm Hg, 30 °C). The resultant green oil (Figure 5B) is then loaded onto a chromatography column (39 cm height x 6 cm I.D.) containing silica gel (170 g) (Note 20). The product is acid sensitive and can decompose during silica gel chromatography, so it is important to flush the column with 300 mL of 5% triethylamine in hexane (Notes 21 and 32) before loading the product on the column. The column is eluted with a 50% mixture of DCM in hexane (Notes 12 and 21) in 50 mL fractions (Note 33). The fractions are checked for product by TLC (Figure 5C) (Note 34). Fractions 4-15 contained the desired product by TLC. These fractions are combined in a 1-L round bottom flask, and the solvent is removed via rotary evaporation (<1 mm Hg, 30 °C) in portions. The resulting light brown oil is transferred to a tared 20-mL scintillation vial using DCM (10 mL). The solvent is again removed via rotary evaporation, and the product is dried under vacuum (<1 mm Hg) overnight to give a brown solid (Figure 5D).
The product is further purified by recrystallization. The solid is transferred to a 100-mL Erlenmeyer flask and dissolved in 50 mL boiling diethyl ether (Note 13). The flask is covered with a small amount of aluminum foil and allowed to cool to room temperature, then cooled to -20 °C in a freezer overnight. The resulting white crystals are collected by suction filtration (300 mmH g) using a Buchner funnel and a 5.5-cm diameter filter paper (No. 1). The crystals are washed twice with 15 mL ice-cold diethyl ether (Note 13). The crystals are transferred to a tared 20-mL scintillation vial and dried under vacuum (<1 mm Hg) for 3 h to give the first crop of 9,9-dibenzyl-1,6-dioxaspiro[4.4]nonan-2-one (3) (2.31 g) as off-white crystals. The mother liquor is transferred to a round bottom flask, and the solvent is removed via rotary evaporation (<1 mm Hg, 30 °C). The resulting brown oil is transferred to a 50-mL Erlenmeyer flask and taken up in 20 mL of boiling diethyl ether (Note 13). The solution is seeded with approximately 5 mg of the previously obtained crystal. The solution is cooled to room temperature and then to -20 °C in a freezer overnight. The resulting white crystals are collected via suction filtration (300 mgH g) using a Buchner funnel and a 5.5-cm diameter filter paper (No. 1). The crystals are washed twice with 15 mL ice-cold diethyl ether (Note 13). The crystals are transferred to a tared 20-mL scintillation vial and dried under vacuum (<1 mm Hg) overnight to give the second crop of compound 3 (0.20 g) as off-white crystals. The first and the second crops are combined to give compound 3 (2.51 g, 7.79 mmol, 73%) (Note 35) (Figure 5E).

Figure 5. A. The celite and silica plug; B. The crude product; C. The TLC of the column chromatography fractions; D. The product after the column; E. The crystalized product (Photos A, B, C and E provided by authors, photo C provided by checkers)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
hexamethyldisilazane,
n-butyllithium,
benzyl bromide,
tetrahydrofuran,
titanium (IV) isopropoxide,
methyl magnesium bromide,
ethyl magnesium bromide,
palladium (II) acetate,
2,3-dichloro-5,6-dicyano-1,4-benzoquinone,
benzene,
dichloromethane,
diethyl ether,
hexane,
ethyl acetate, silica gel, and deuterated chloroform. Some of these procedures call for the use of a benzophenone ketyl still to dry solvents. Benzophenone ketyl stills must be properly used and maintained in order to mitigate the risk of fire. Organometallic reagents, such as
n-butyllithium,
methyl magnesium bromide, and
ethyl magnesium bromide are extremely flammable and corrosive and react vigorously with moisture. Care should be taken to ensure that these compounds do not come into contact with any water or air. Chronic exposure to
benzene has been linked to cancer in humans;
benzene should always be handled in a fume hood.
2. The room temperature throughout this manuscript refers to temperature between 22 °C and 25 °C.
3.
Bis(trimethylsilyl)amine (99%) was purchased from Sigma Aldrich and was used as received.
4.
THF (≥99.9%) was purchased from Kanto Chemical CO., INC. (https://www.kanto.co.jp) and was dried using a GlassContour Solvent Purification System from Nikko Hansen.
5. For all reactions, a Yazawa magnetic stirrer (model KF-82MR) was used.
6.
n-Butyllithium (2.3 M solution in cyclohexane, Sure/Seal
TM packaging system) was purchased from Tokyo Chemical Industry Co., Ltd. and used as received.
7.
γ-Butyrolactone (≥99%) was purchased from Sigma Aldrich and used as received.
8.
Benzyl bromide (>98%) was purchased from Tokyo Chemical Industry Co., Ltd. and used as received.
9.
Ethyl acetate (>99%) was purchased from NACALAI TESQUE, INC. and used as received.
10.
Sodium sulfate (>98.5%) was purchased from NACALAI TESQUE, INC. and used as received.
11. Celite
® 545 was purchased from FUJIFILM Wako Pure Chemical Corporation and was used as received.
12.
Dichloromethane (≥99.5%) was purchased from JUNSEI and used as received.
13.
Diethyl ether (≥99%) was purchased from Showa Ether, INC. and used as received.
14.
3,3-Dibenzyldihydrofuran-2(3H)-one (1) has the following physical properties: m.p. 133-135 °C; IR (neat): 3025, 2922, 1752, 1494, 1455, 1377, 1222, 1168, 1082, 1027, 761, 701;
1H NMR
pdf (400 MHz, CDCl
3) δ 7.37 - 7.17 (m, 10H), 3.37 (t,
J = 7.4 Hz, 2H), 3.21 (d,
J = 13.4 Hz, 2H), 2.79 (d,
J = 13.5 Hz, 2H), 2.16 (t,
J = 7.4 Hz, 2H);
13C NMR
pdf (101 MHz CDCl
3) δ 181.1, 136.4, 130.1, 128.6, 127.1, 65.2, 49.8, 43.8, 29.0; HRMS (ESI):
m/z calc'd for C
18H
19O
2 [M+H]
+: 267.1380. Found: 267.1373. The purity was determined to be 98 wt% by qNMR
pdf, using
1,3,5-trimethoxybenzene (Sigma-Aldrich, >99%) as the internal standard.
15.
THF (≥99.9%) was purchased from Sigma Aldrich was dried using sodium benzophenone still.
16.
Ti(Oi-Pr)4 (97%) was purchased from Sigma Aldrich and used as received.
17.
MeMgBr, 3 M solution in
diethyl ether was purchased from Sigma Aldrich and used as received.
18.
EtMgBr 3 M solution in
diethyl ether was purchased from Sigma Aldrich and used as received.
19. The reaction was traced via TLC and was quenched when the TLC no longer showed signs of the starting lactone. To take the TLC, a stopper was removed from the reaction flask, and a glass pipette was dipped into the reaction mixture so that tip contained about 0.1 mL of the reaction mixture. The reaction stopper was replaced, and the pipette was rinsed into a 1-dram vial with 0.5 mL of
ethyl acetate. The organic layer was quenched with 0.5 mL of water. The organic layer was spotted onto a TLC plate (60 F
254 glass plates precoated with a 0.25 mm or thickness of silica gel). The TLC plate was run in a 25% solution of
ethyl acetate in
hexane. The TLC was stained in
KMnO4. In this system the product has an Rf of 0.13. In the image, the left spot contains starting material, the center spot is a co-spot of starting material and the reaction mixture, the right spot is only the reaction mixture.

Figure 6. Reaction TLC; starting material (left), reaction mixture (right), and co-spot (center) (photo provided by checkers)
20. Silica gel (Silica gel 60 N, 0.040-0.050 mm, spherical and neutral) was purchased from Kanto Chemical CO., INC. and used as received.
21.
Hexane (≥98.5%) was purchased from FUJIFILM Wako Pure Chemical Corporation and used as received.
22. The fractions were spotted onto a TLC plate (60 F
254 glass plates precoated with a 0.25 mm or thickness of silica gel). The TLC plate was run in a 25% solution of
ethyl acetate in
hexane. The TLC was stained in cerium ammonium molybdate.
23. A
1,3,5-Trimethoxybenzene (Sigma Aldrich, >99%) internal standard was used to check the purity of combined fractions (19-25) and (13-19 + 26-32) by qNMR
pdf pdf.
24.
1-(2-Benzyl-4-hydroxy-1-phenylbutan-2-yl)cyclopropan-1-ol (2) has the following characteristics. IR (neat): 3400 (br), 3028 (s), 2935 (s), 1602 (s), 1495 (s), 1454 (s), 1183 (s), 1031 (s), 750 (s), 702 (s);
1H NMR
pdf (400 MHz, CDCl
3) δ 7.32 -7.29 (m, 4H), 7.28 -7.22 (m, 6H), 4.00 (t,
J = 6.0 Hz, 2H), 2.74 (d,
J = 13.2 Hz, 2H), 2.31 (d,
J = 13.2 Hz, 2H), 1.79 (t,
J = 6.0 Hz, 2H), 0.83 - 0.80 (m, 2H), 0.68 - 0.65 (m, 2H).;
13C NMR
pdf (101 MHz CDCl
3) δ 138.4, 130.7, 128.2, 126.3, 58.7, 58.7, 43.6, 41.9, 35.0, 10.2; HRMS (ESI):
m/z calc'd for C
20H
25O
2 [M+H]
+: 297.1849. Found: 297.1819.
25.
Benzene (≥99.7%) was purchased from Sigma Aldrich and dried in a sodium benzophenone still before use.
26.
DDQ (98%) was purchased from Sigma Aldrich and used as received.
27. Excess
DDQ is used in this reaction to ensure the cycling of the palladium catalyst and avoid the production of palladium black.
28.
Carbon monoxide (99%) was purchased from Taiyo Nippon Sanso and was used as received.
29. For each cycle, the vacuum was maintained for at least 3 min to ensure full degassing of the solution.
30.
Palladium acetate (98%) was purchased from Sigma Aldrich and used as received.
31. The reaction was traced via TLC and was quenched when the TLC showed trace amounts of the cyclopropanol. The reaction was spotted onto a TLC plate (60 F
254 glass plates precoated with a 0.25 mm or thickness of silica gel). The TLC plate was run in a 25% solution of
ethyl acetate in
hexane. The TLC was stained in cerium ammonium molybdate stain. In this system the product has an Rf of 0.42.
32.
Triethylamine (99%) was purchased from FUJIFILM Wako Pure Chemical Corporation and used as received.
33. The first fraction of the column was 200 mL and the remaining fractions were 50 mL.
34. The fractions were spotted onto a TLC plate (60 F
254 glass plates precoated with a 0.25 mm or thickness of silica gel). The TLC plate was run in a 50% solution of
dichloromethane in
hexane. In this system the TLC has an Rf of 0.11. The TLC was stained in cerium ammonium molybdate.
35.
9,9-Dibenzyl-1,6-dioxaspiro[4.4]nonan-2-one (
3) has the following physical properties: m.p. 108-109 °C; IR (neat): 3060 (s), 3028 (s), 2948 (s), 2899 (s), 1775 (s), 1496 (s), 1456 (s), 1268 (s), 1011 (s), 969 (s), 755 (s), 703 (s);
1H NMR
pdf (400 MHz, CDCl
3) δ 7.33-7.26 (m, 10H), 3.97 (dd,
J = 5.2, 4.8 Hz, 1H), 3.62 (dt,
J = 8.4, 8.4 Hz, 1H), 3.13 (d,
J = 14.0 Hz, 1H), 2.99 (d,
J = 14.4 Hz, 1H), 2.83 (d,
J = 14.0 Hz, 1H), 2.67 (ddd,
J = 17.6, 10.4, 10.4 Hz, 1H), 2.57 (d,
J = 14.0 Hz, 1H), 2.19 (dd,
J = 18.0, 10.4 Hz, 1H), 2.11 - 2.03 (m, 2H), 1.98 (dd,
J = 14.0, 8.0 Hz, 1H), 1.68 (dt,
J = 14.0, 10.4 Hz, 1H);
13C NMR
pdf (100 MHz, CDCl3) δ 176.3, 137.4, 137.3, 130.6, 130.5, 128.3, 128.3, 126.7, 126.7, 118.9, 65.9, 51.7, 39.3, 37.0, 33.6, 28.4, 27.2; HRMS (ESI):
m/z calc'd for C
21H
23O
3 [M+H]
+: 323.1642. Found: 323.1656. The purity was determined to be 99 wt% by qNMR
pdf, using
1,3,5-trimethoxybenzene (Sigma-Aldrich, >99%) as the internal standard.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Oxaspirolactones appear frequently in natural products and synthetic molecules with biological activities.
2 Their prevalence means that methods of synthesizing these moieties have become very important in the synthetic community. This procedure is a mild and efficient way of forming these oxaspirolactones from hydroxycyclopropanols. This method has already shown use in the total synthesis of the prostaglandin D
2 metabolite (PGDM) methyl ester.
3 This is our second-generation catalytic carbonylative spirolactonization
3,4. The first-generation method has a broad substrate scope
5 and has been in used in the synthesis of bisdehydroneostemoninine, bisdehydrostemoninine, tuberostemoamide, sessilifoliamide A, α-levantanolide, and α-levantenolide.
5,6,7 However, the first generation uses longer reaction times, higher temperatures, and an expensive ($1,961.00/g, AmBeed) dimeric, monocationic palladium catalyst, [Pd(neoc)(OAc)]
2(OTf)
2, which require 3 steps to prepare.
8 The second generation is more scalable and user friendly as it is run at room temperature within a few hours and uses the much more available palladium acetate. The first and second generations have comparable yields at 0.1 and 0.2 mmol scales (Figure 6).
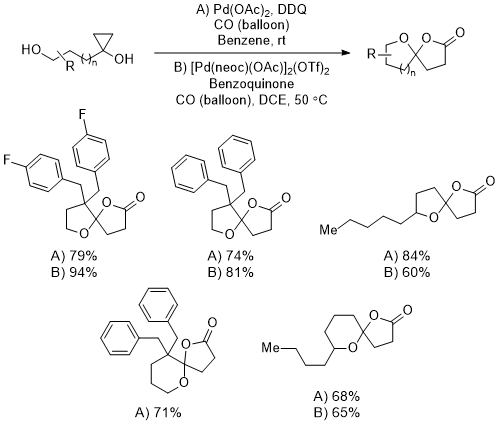
Figure 7. The comparison between the first- and second-generation methods at smaller scales. The yields from the second-generation are marked with A, and the first generation are marked with B
Appendix
Chemical Abstracts Nomenclature (Registry Number)
Hexamethyldisilazane: 1,1,1-trimethyl-N-(trimethylsilyl)-silanamine; (CAS 999-97-3)
n-Butyllithium: Butyllithium; (109-72-8)
Benzyl Bromide: (Bromomethyl)-benzene; (100-39-0)
γ-Butyrolactone: Dihydro-2(3H)-Furanone; (96-48-0)
Ti(OiPr)4: Titanium(4+) salt (4:1) 2-Propanol; (546-68-9)
MeMgBr: Bromomethyl magnesium; (75-16-1)
EtMgBr: Bromoethyl magnesium; (925-90-6)
Palladium Acetate: Palladium(2+) salt (2:1) acetic acid; (3375-31-3)
DDQ: 4,5-Dichloro-3,6-dioxo-1,4-cyclohexadiene-1,2-dicarbonitrile; (84-58-2)

|
Cyrus Gudeman was born was born and raised in Morton, IL. In 2021 he received his BS in chemistry from Illinois State University where he carried out research under the direction of Professor Timothy Lash. In 2021 he moved to Purdue University and began work in Professor Mingji Dai's laboratory. In 2022 the Dai lab moved to Emory University, where Cyrus is currently a fourth-year graduate student. His studies focus on synthetic methodology development and natural product total synthesis. |

|
Courtney Wiethorn was born and raised in Waco, TX. In 2022, she began her undergraduate studies at Emory University as a Chemistry major. Courtney joined professor Mingji Dai's laboratory in the fall of 2023. She is currently a sophomore student. Her research focuses on synthetic methodology development and natural product total synthesis. |

|
Mingji Dai received his B.S. degree in 2002 from Peking University under the guidance of Professors Zhen Yang and Jiahua Chen and earned his Ph.D. degree in 2009 under the guidance of Professor Samuel J. Danishefsky at Columbia University. He then took a postdoctoral position in the laboratory of Professor Stuart L. Schreiber at Harvard University and the Broad Institute. In 2012, he began his independent career at Purdue University and was promoted to full professor in 2020. In August 2022, he moved to Emory University and is currently the Asa Griggs Candler Professor of Chemistry. |

|
Yusuke Kanno was born in Sendai, Japan in 2000. He received his B.S. (2023) and M.S. (2025) from Tohoku University (Japan) under the supervision of Professor Hidetoshi Tokuyama. He is currently pursuing his Ph.D. at the same graduate school. His research interests are the area of the total synthesis of complex natural products. |

|
Juri Sakata was born in Shizuoka, Japan in 1986, and received his BSc (2009), MSc (2011) from Kogakuin University under the supervision of Professor Shinji Nagumo and Professor Masaaki Miyashita. He then moved to the laboratories of Professor Keisuke Suzuki at the Tokyo Institute of Technology and got his Ph. D. in 2015. In 2015, he joined the group of Professor Hidetoshi Tokuyama and was appointed Assistant Professor. His current research interest is total synthesis of complex natural product. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved