Org. Synth. 2026, 103, 19-34
DOI: 10.15227/orgsyn.103.0019
Base-Promoted N-Heteroaryl C-H Oxidative Coupling With Alcohols for N-Heteroaryl Ether Synthesis
Submitting Authors: Samuel L. Graf
1 and Jeffrey S. Bandar
1*
Checking Authors: Partha Sarathi Hazra and M. Kevin Brown*
21. Procedure (Note 1)
A. 4-(Cyclopropylmethoxy)-2,6-bis(trifluoromethyl)pyridine (4). An oven-dried 500-mL single-neck (24/40 joint), round-bottom flask is charged with an oven-dried PTFE-coated magnetic stir bar (4.0 cm x 1.0 cm.), 2,6-bis(trifluoromethyl)pyridine (1) (5.00 g, 23.2 mmol, 1.0 equiv) (Note 2), cyclopropanemethanol (2) (2.52 g, 34.9 mmol, 1.5 equiv) (Note 3), 2-iodothiophene (3) (9.77 g, 46.5 mmol, 2.0 equiv.) (Note 4), and 18-crown-6 ether (9.22 g, 34.9 mmol, 1.5 equiv) (Note 5). The flask is sealed with a rubber septum, a needle from a manifold Schlenk line is inserted, the flask is evacuated and then backfilled with N2 three times and left under positive pressure of N2. Anhydrous THF (186 mL) (Note 6) is added through the septum via a syringe (50-mL) equipped with a 22G needle and the reaction mixture is placed in an ice bath and allowed to cool to 0°C with stirring set to 800 RPM (Figure 2a).
A separate oven-dried 100-mL round-bottom (24/40 joint) flask is charged with an oven-dried magnetic stir bar (2 cm. x 0.65 cm) and KO-t-Bu (4.207 g, 37.5 mmol) (Note 7). The flask is sealed with a rubber septum, a needle from a manifold Schlenk line is inserted, the flask is evacuated and then backfilled with N2 three times and left under positive pressure of N2. Anhydrous THF (50 mL) is added through the septum via a syringe (50-mL) equipped with a 22G needle and the mixture is stirred for approximately 1 min until homogenous (Figure 1). The 0.75 M KO-t-Bu in THF solution (46.5 mL, 34.9 mmol, 1.5 equiv.) is then added via syringe (50-mL) equipped with a 22G needle over 10 min to the round-bottom flask containing 2,6-bis(trifluoromethyl)pyridine (1), cyclopropanemethanol (2), 2-iodothiophene (3), and 18-crown-6 ether in THF while stirring at 0°C. The resulting reaction mixture begins to turn dark orange, forming a precipitate after approximately 30 min (Figure 2c).

Figure 1: KO-t-Bu in THF (0.75M) solution under N2
Figure 2: Reaction mixture shown at various time points; A. Arene and alcohol starting materials under N2 in THF at 0°C; B. Reaction mixture immediately following base addition; C. 30 min time point, salt starts to precipitate; D. 1 h time point, ice bath removed; E. 2 h time point; F. 4 h time point; G. 6 h time point
The reaction flask is removed from the ice bath after 30 min and allowed to warm to room temperature (20°C) with continued stirring at 800 rpm for a total time of 6 h, during which time additional KI precipitate forms from the reaction mixture (Figures 2d-2g).
Figure 3: Reaction workup process; A. Separatory funnel with 200 mL DI H2O; B. First extraction of crude mixture with 200 mL of EtOAc; C. Aqueous and organic extracts; D. Organic extracts dried with Na2SO4; E. Gravity filtration to remove Na2SO4; F. Crude extract; G. Concentrated crude extract
After 6 h, deionized water (30 mL) is added to the reaction mixture and stirred to dissolve the precipitate before the biphasic mixture is transferred to a 1-L separatory funnel containing additional water (200 mL) (Figure 3a). The aqueous layer is extracted with EtOAc (3 x 200 mL) (Figure 3b). Subsequently, the combined organic extract is washed with brine (50 mL) and dried with anhydrous Na2SO4 (150 g) (Note 8) (Figure 3d). The Na2SO4 is removed via gravity filtration (Whatman Grade 1 Qualitative 185 mm diameter filter paper) (Figure 3e) to a 1-L round-bottom flask (Figure 3f). The filtrate is concentrated via a rotary evaporator to remove the solvent and other volatile materials (40°C, 40 mmHg). This results in a viscous brown oil (Figure 3g). A TLC (Note 9) of this material compared to relevant starting materials and byproducts is shown in Figure 4.
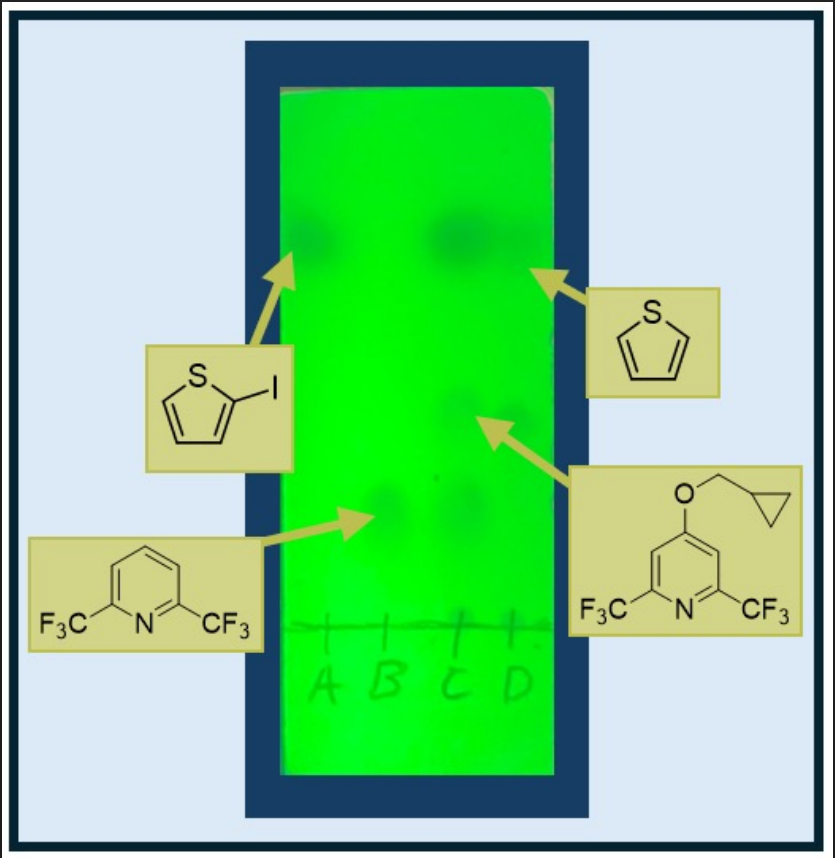
Figure 4. Visualization of TLC of crude reaction mixture under UV lamp (λmax = 254 nm) (2% ethyl acetate in hexanes eluent mixture); A. 2-iodothiophene (3), R f = 0.85; B. 2,6-bis(trifluoromethyl)pyridine (1), Rf = 0.21; C. Co-spot, 4-(cyclopropylmethoxy)-2,6-bis(trifluoromethyl)pyridine (4), Rf = 0.62; D. Crude Reaction Mixture, thiophene, Rf = 0.73
The resulting crude material is purified via flash column chromatography. The column (6.35 cm x 30.5 cm) is loaded with dry silica gel (24 cm) (Note 10) and flushed with 1 L of hexanes (Note 11). The crude material is then concentrated on silica gel (15 g) with ethyl acetate (Note 12) via the rotary evaporator (40°C, 40 mmHg). This mixture is loaded on the column before sea sand (1 cm) (Note 13) is layered on top (Figure 5A). The column is eluted using air pressure with a gradient starting with 200 mL of hexanes followed by 400 mL of 1% EtOAc/hexanes, and 2 L of 2% EtOAc/hexanes, which were discarded. The column is sequentially charged with an additional 2 L of 2% EtOAc/hexanes and the fractions are collected (18 x 150mm) test tubes, (20 mL fractions). The eluent containing the product (Fractions 10-34) is combined into a 2-L round-bottom flask (Figure 5B-D) and concentrated via rotary evaporation (40°C, 40 mmHg).

Figure 5: A. Column with dry-loaded sample after flushed with 200 mL of hexanes; B. Column midway through elution; C. Column at the end of elution; D. Visualization of TLC of column fractions under UV lamp (λmax = 254 nm) (2% ethyl acetate in hexanes eluent mixture)
The concentrated product is transferred to a 20-mL scintillation vial and dried under high vacuum (0.1 mm Hg) overnight (16 h) to yield a clear, pale-yellow liquid (5.46 g, 82% yield, 98.1% purity determined by qNMR analysis) (Figure 6, Notes 14,15,16,17 and 18). A second run on the same scale provided 79% isolated yield with 98.1% purity.
Figure 6: Isolated product
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available
via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
2,6-bis(trifluoromethyl)pyridine (
1),
cyclopropanemethanol (
2),
2-iodothiophene (
3),
18-crown-6 ether,
potassium tert-butoxide,
tetrahydrofuran,
ethyl acetate and hexanes, as well as the proper procedures for flash chromatography.
2.
2,6-Bis(trifluoromethyl)pyridine (97%) was purchased from Ambeed and used as received.
3.
Cyclopropanemethanol (98%) was purchased from MilliporeSigma and used as received.
4.
2-Iodothiophene (98%) was purchased from Ambeed and used as received.
5.
1,4,7,10,13,16-Hexaoxacyclooctadecane (
18-crown-6 ether) (>99.5%) was purchased from Chem-Impex and used as received.
6.
Tetrahydrofuran stabilized with 0.025% butylated hydroxytoluene was purchased from Fisher Chemical and was deoxygenated and dried by passage over packed columns of neutral alumina and copper (II) oxide under positive pressure of
N2 before use.
7.
Potassium tert-butoxide (99%) was purchased from MilliporeSigma and used as received.
8. Granular anhydrous
sodium sulfate (>99%) was purchased from Fisher Chemical and used as received.
9. Silica gel 60Å F
254plates (250 µm, SiliaPlate) were purchased from Silicycle and used as received.
10. SiliFlash® P90 40-63 µm 60Å silica gel was purchased from Silicycle and used as received.
11. Hexanes (98.5%) was purchased from Fisher Chemical and used as received.
12.
Ethyl acetate (>99.5%) was purchased from Fisher Chemical and used as received.
13. Sea sand was purchased from Fisher Chemical and used as received.
14.
Trimethoxybenzene (98%) was purchased from Ambeed and used as received.
15. Given that the reaction produces a KI salt precipitate as a byproduct, it is noted that if the reaction mixture is of higher concentration, or if the rate of stirring is insufficient, or if the solution is not cooled prior to addition, the precipitate may form rapidly and cause the entire mixture to turn into a gel-like suspension. This suspension can be broken through agitation of the mixture.
16. The reaction is mostly complete after 2 h (~70% conversion) with additional time allowing the reaction to go to completion. This is desirable as the arene starting materials may coelute with the product. TLC analyses were performed using 2%
EtOAc/hexanes as eluent and visualization was performed with a 254 nm UV lamp. Glass backed TLC silica gel plates were purchased from Silicycle. R
f-value of
2,6-bis(trifluoromethylpyridine) = 0.21, R
f-value of
4-(cyclopropylmethoxy)-2,6-bis(trifluoromethyl)pyridine = 0.62, R
f-value of
2-iodothiophene = 0.85. After 6 h, minimal starting material remains.
17.
4-(Cyclopropylmethoxy)-2,6-bis(trifluoromethyl)pyridine (
4) has the following spectroscopic properties:
1H NMR
pdf (500 MHz, CDCl
3) δ 7.31 (s, 2H), 3.99 (d,
J = 7.1 Hz, 2H), 1.36 - 1.25 (m, 1H), 0.77 - 0.66 (m, 2H), 0.40 (dt,
J = 6.5, 4.8 Hz, 2H).
13C NMR
pdf (126 MHz, CDCl
3) δ 167.8, 150.6 (q,
J = 35.6 Hz), 121.3 (q,
J = 274.7 Hz), 110.2 (q,
J = 2.8 Hz), 74.7, 9.9, 3.7.
19F NMR
pdf (471 MHz, CDCl
3) δ -68.33. HRMS (ESI) m/z Calcd. for [M+H] C
11H
10F
6NO 286.0661; found 286.0656. FTIR
(ν, cm
-1): 3094, 3015, 2891, 1614, 1583, 1463, 1423, 1394, 1346, 1274, 1192, 1148, 1085, 1029, 1013, 930, 892, 869, 735, 695.
18. The purity of 98.1 wt % was determined by qNMR
pdf using
1,3,5- trimethoxybenzene as internal standard.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654 ). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc. , its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
1,3-Azole- and (di)azine-based heteroaryl ethers are key structural moieties in many useful products, including pharmaceuticals and agrochemicals.
3,4 The most common method for preparing these products is through base-promoted S
NAr reactions or metal-catalyzed C-O coupling reactions with alcohol pronucleophiles.
5,6 However, a requirement for these approaches is the use of
N-heteroaryl halide coupling partners. A direct C-H alcohol coupling reaction would provide an attractive route to
N-heteroaryl ethers in cases where the corresponding
N-heteroaryl halide is not commercial, is expensive, or is not easily prepared. In this regard, we developed a base-promoted direct oxidative coupling reaction of
N-heteroaryl C-H bonds using 2-halothiophenes as a new class of halogen oxidants.
7 This protocol complements alternative multistep routes for net C-H etherification, including traditional metalation/halogenation sequences,
8 N-oxide activation,
9 and McNally's pyridine phosphonium salt platform.
10The protocol described above is proposed to operate
via a base-promoted halogen transfer sequence that is shown in Figure 7. The key step of this process, aromatic halogen transfer between a 2-halothiophene and an
N-heteroaryl carbanionic intermediate, is inspired by the mechanism of classical halogen dance methodology.
11 Such aromatic halogen transfer processes are not often controllable unless a strong thermodynamic driving force is provided.
12 We realized that aromatic halogen transfer could be driven towards completion when sequenced with a rapid S
NAr step. Thus, 2-halothiophenes serve as base-compatible oxidants and allow for net C-H aromatic substitution reactions under the basic reaction conditions that are often used in traditional S
NAr protocols.
Figure 7: Proposed mechanism of oxidative etherification via aromatic halogen-transfer substitution
This method is applicable to diverse
N-heteroaryl substrates by varying the sacrificial 2-halothiophene oxidant, denoted as halogen transfer reagents (XTRs). Inexpensive 2,5-dibromothiophene is an effective oxidant for 1,3-azole substrates. A switch to 2-iodothiophene (also inexpensive) is required for (di)azines; this is likely because (di)azines are generally less acidic than 1,3-azoles, meaning that a lower concentration of aryl carbanionic intermediates are generated at any given time and thus a faster electrophilic halogen transfer process is required.
13 In this regard, iodine is known to be more labile than bromine for halogenophilic processes.
14 In certain cases, competing disproportionation 2-iodothiophene can result in decreased yield, an issue that can be resolved using 2,3-diiodobenzothiophene. Use of these reagents enables highly regioselective etherification of diverse classes of
N-heteroarenes with a p
Ka up to approximately 38 in DMSO.
13 Representative products are shown in Figure 8. These illustrate that functionalization generally occurs at the most acidic and S
NAr-active aryl position of the substrate (e.g., 2-position of 1,3-azoles and 4-position of pyridines), although a more nuanced discussion of these considerations is provided in a recent article.
15
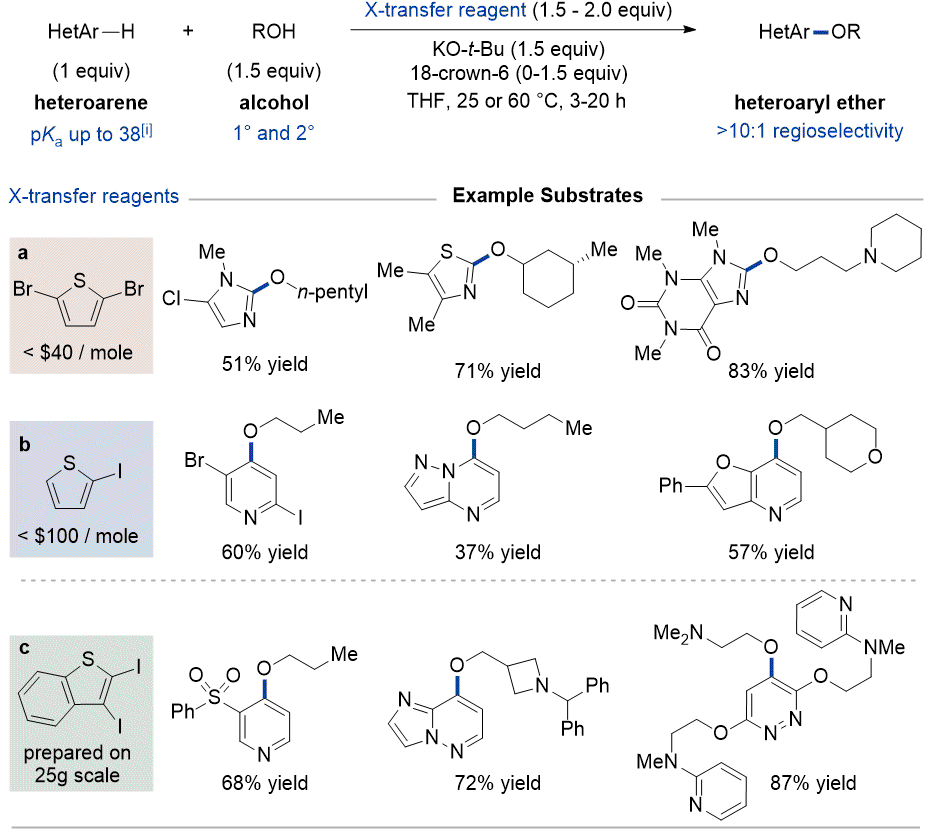
Figure 8: Representative C-H etherification substrate scope (For full scope see ref 7 ), [i] pKain DMSO (ref 13)
The procedure detailed herein is slightly altered from the original publication
7 to make it suitable for a robust and reproducible multigram scale synthesis. When all reagents are added rapidly at room temperature, a slight exotherm is noted. Thus, addition of base to an ice-bath-cooled solution of reactants was used herein to mitigate any safety concerns. Furthermore, we found that under the 0.5 M concentration conditions of our initial report, reactions of fast-reacting substrates (including compound
1) generate large amounts of KI precipitate that solidifies the entire reaction mixture. By reducing the concentration to 0.1 M and adding the KO-
t-Bu solution last, we established a more robust procedure that generates precipitate in a controlled manner (see Figure 2).
In summary, the protocol described herein provides a practical and inexpensive method for
N-heteroaryl C-H etherification. In recent advances, we have shown that this strategy is also applicable to less acidic benzene substrates and that the use of 2-phenylethanol enables direct aryl C-H hydroxylation reactions.
15 More broadly, the mechanistic platform described in Figure 7 is also being expanded to other classes of C-H bonds and nucleophiles, including for benzylic C-H oxidative coupling reactions.
16
Appendix
Chemical Abstracts Nomenclature (Registry Number)
2,6-bis(trifluoromethyl)pyridine (1) (455-00-5)
cyclopropanemethanol (2) (2516-33-8)
potassium tert-Butoxide (865-47-4)
18-crown-6 ether (1,4,7,10,13,16-hexaoxacyclooctadecane) (17455-13-9)
2-iodothiophene (3) (3437-95-4)

|
Samuel Graf was born in California in 2001 and graduated with a B.S. in Chemistry from the University of Florida in 2024. He is pursuing his Ph.D. at Colorado State University in Fort Collins working under Dr. Jeff Bandar. His research focuses on expanding halogen transfer reactivity and applications. |

|
Jeff Bandar received his B.A. in Chemistry from Saint John's University in Collegeville, Minnesota in 2009. He then conducted Ph.D. studies with Tristan H. Lambert at Columbia University from 2009 to 2014, followed by postdoctoral work with Stephen L. Buchwald at Massachusetts Institute of Technology. In 2017, Jeff started his independent research career at Colorado State University where his group focuses on the discovery and development of new base-promoted synthetic methods. |

|
Partha Sarathi Hazra was born in Midnapore, West Bengal, India. He received his Bachelor of Science (B.Sc. Hons) in Chemistry from Midnapore College (Autonomous) and Master of Science (M.Sc.) from Indian Institute of Technology (IIT)-Bombay, where he worked with Prof. Debabrata Maiti in C-H activation. Upon completion of his master's study, he moved to Indiana for perusing Ph.D. under the supervision of Prof. M. Kevin Brown at Indiana University-Bloomington in 2022. |

|
M. Kevin Brown grew up in the suburbs of Chicago. Kevin received his B.A. degree from Hamilton College in 2002 and then moved to Boston College to pursue graduate studies under the mentorship of Professor Amir Hoveyda. Upon completion of his graduate studies, he began a postdoctoral fellowship in the laboratories of E.J. Corey at Harvard University. In 2011 he began his independent career at Indiana University as an assistant professor and was full professor in 2020. His research interests are focused on the development of new methods for chemical synthesis. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved