Org. Synth. 2026, 103, 64-77
DOI: 10.15227/orgsyn.103.0064
Amination of Aryl Boronic Acids Using O-(Diphenylphosphinyl)hydroxylamine (DPPH)
Submitted by Kasie H. Leung and Richard Y. Liu*
1Checked by Kanaram Kumawat and M. Kevin Brown*
21. Procedure (Note 1)
4-Amino-N-(phenylmethyl)benzamide (1). A 500-mL, three-necked (24/40 joint) round-bottomed flask was equipped with a 1.5-inch elliptical stir bar, two rubber septa, and a glass inlet adaptor (Note 2). The flask was connected to a manifold, placed under vacuum, flame-dried, and then cooled under N2. A septum was removed, and the flask was charged with (4-(benzylcarbamoyl)phenyl)boronic acid (7.65 g, 30 mmol, 1.0 equiv) and O-(diphenylphosphinyl)hydroxylamine (DPPH) (Note 3) (8.40 g, 36 mmol, 1.2 equiv) (Figure 1) (Note 3). The flask was resealed with the septum and brought under inert atmosphere by evacuating and back-filling with N2 for a total of 3 cycles. Through a septum, tetrahydrofuran (THF, 100 mL) (Note 4) was added via syringe (50-mL syringe, 22-gauge needle), the reaction mixture was stirred with a magnetic stirrer (500 RPM), and the flask was placed in an ice-water bath. After 10 min, a septum was temporarily removed, and freshly ground potassium hydroxide (KOH) (5.05 g, 90 mmol, 3.0 equiv) was added portion-wise over the course of one minute (Note 5) such that no boiling of the solvent was observed (Note 6). After the addition of KOH, the reaction mixture was a light-yellow slurry. The flask was removed from the ice-water bath and allowed to warm to room temperature (25 °C).

Figure 1: Reaction assembly after A. exchanging atmosphere with nitrogen; B. addition of THF and cooling in ice bath; and C) 2 h of reaction time
The reaction progress was monitored by TLC (Note 7, 8). After 30 min, boronic acid remained, and addition of a second portion of DPPH (3.49 g, 15 mmol, 0.5 equiv) was performed in the same manner as the first addition (Note 9). The stir rate was increased to 800 RPM, and after a further 90 min, the boronic acid was consumed as judged by TLC analysis. Upon completion, the reaction mixture appeared as a yellow solution with significant white precipitate. The reaction mixture was vacuum-filtered through a 150-mL coarse-porosity sintered glass funnel into a 500 mL round-bottomed flask. To ensure quantitative transfer, the reaction flask was washed with THF (3 x 5 mL). The filtrate was concentrated with the aid of a rotary evaporator (35 °C, 60 mmHg) to afford a heterogeneous beige mixture.
Figure 2: Reaction mixture after A. filtration; B. removal of THF
The crude mixture was diluted with ethyl acetate (100 mL) and sonicated for ca. 1 min to suspend the solids. 1 M hydrochloric acid (30 mL) was added to the flask and the mixture was vacuum filtered through a 150-mL coarse-porosity sintered glass funnel into a 250-mL separatory funnel using a 24/40 straight vacuum adaptor. The phases were separated, and product was extracted from the orange organic layer with 1 M hydrochloric acid (9 x 30 mL), combining the fractions into a 500 mL Erlenmeyer flask (Note 10).
Figure 3: Partition of acidic aqueous and organic layers during A. extraction of product; B. ethyl acetate wash; C. recovery of product from combined organic layers used for the ethyl acetate wash
The combined acidic layers were transferred to a 500-mL separatory funnel and washed with ethyl acetate (3 x 50 mL) (Note 11). The yellow organic layers used for washing were combined in a 250 mL separatory funnel, and the product was back-extracted with 1 M hydrochloric acid (6 x 10 mL) (Note 10). All aqueous layers were combined in a 500-mL Erlenmeyer flask, which was then equipped with a 1.5-inch elliptical magnetic stir bar, placed in an ice-water bath, and neutralized to pH 7 (Note 12) by portion-wise addition of ca. 22 g of sodium bicarbonate while stirring at 300 RPM. The formation of a white precipitate was observed upon neutralization.
Figure 4: Combined aqueous layers after A. neutralization; B. back-extraction and addition of MgSO4; C. removal of dichloromethane
The neutralized aqueous layer was transferred to a 500-mL separatory funnel, and the product was extracted with dichloromethane (6 x 50 mL). The combined organic layers were washed with saturated aqueous NaCl solution (50 mL) and dried over 15 g of anhydrous Na2SO4 (Note 13). The solution was filtered through a 60-mL polyethylene frit funnel (Note 14), rinsing the Erlenmeyer flask with dichloromethane (3 x 10 mL). The pale-yellow filtrate was concentrated in a 500-mL flask with the aid of a rotary evaporator (60 mmHg, 35 °C) to afford a pale-yellow powder (Note 15).
Figure 5: Recrystallization solution after A. addition of hexanes; B. further addition of isopropanol; C. cooling to 25 °C
To recrystallize, the flask was equipped with a 1.5-inch elliptical stir bar and a reflux condenser. The crude mixture was dissolved in isopropanol (10 mL) by heating to 85 °C in an oil bath while stirring at 300 RPM. Using a Pasteur pipette, hexanes (ca. 12 mL) (Note 16) were added to the clear yellow solution until a persistent cloudiness developed. An additional 1 mL of isopropanol was added, after which the solution became homogenous again. The heat source was turned off, and the flask was allowed to cool to 25 °C in the oil bath with stirring. The resulting pale yellow crystals in a yellow solution were collected in a 150-mL coarse-porosity sintered glass funnel by vacuum filtration. The flask was rinsed with 1:1 isopropanol/hexanes (3 x 3 mL), which was used to wash the filter cake. The solids were transferred to a 20-mL scintillation vial and dried to a constant mass under high vacuum (0.1 mmHg). Compound 1 was obtained as a pale yellow powder (3.52 g, 15.6 mmol, 52% yield) (Note 17,18).

Figure 6: Purified product before and after drying
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/about/governance/committees/chemical-safety.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
[4-(Benzylcarbamoyl)phenyl]boronic acid,
O-(Diphenylphosphinyl)hydroxylamine,
potassium hydroxide,
tetrahydrofuran,
dichloromethane,
methanol,
ethyl acetate,
hydrochloric acid,
sodium bicarbonate,
magnesium sulfate,
isopropanol and hexanes.
2. The joint was greased with Dow Corning high-vacuum grease.
3.
(4-(Benzylcarbamoyl)phenyl)boronic acid was purchased from Ambeed (98%) and used as received.
DPPH (95%) was purchased from Combi-Blocks and used as received.
4. Anhydrous
tetrahydrofuran (≥99.9%, inhibitor-free) was purchased from Sigma-Aldrich. The solvent was degassed by purging with argon and passed through alumina columns prior to use with the aid of a solvent purification system.
5.
KOH was ground in a mortar and pestle directly prior to use, weighed on folded weighing paper and added by removing a rubber septum, while temporarily increasing the flow of
N2 through the adaptor.
Figure 7: Freshly ground KOH in mortar.
6. The authors noted that if the order of addition is reversed (
tetrahydrofuran is added to
KOH) at 25 °C, then a yellow tar is formed in the reaction flask, and the boiling of
tetrahydrofuran can be observed.
7. The reaction mixture was sampled by insertion of a capillary tube through an 18G needle, while stirring was paused.
8. TLC was conducted on glass-backed 60 Å silica gel plates purchased from Sigma-Aldrich with 5%
methanol in
dichloromethane as the mobile phase. UV light (254 nm) was used as the visualization method.
Figure 8: TLC of the reaction mixture (R) versus starting boronic acid (S, Rf = 0.37) with a co-spot (C) taken 30 min, 1h, 1h 30 min, and 2 h after removal from the ice-water bath. The starting material co-elutes with an orange byproduct formed during the reaction
9.
DPPH was added by removing a rubber septum and temporarily increasing the flow of
nitrogen through the glass inlet adaptor.
10. The extraction of the
hydrochloric acid salt of
1 from
ethyl acetate was monitored by TLC of the organic layer.
Figure 9: TLC of purified product (P, Rf = 0.46) versus organic layer after each set of 3 extractions. A. suspension of crude mixture in ethyl acetate; B. combined wash layers. After the extraction is complete, product is not detected in the organic layer
11. The authors noted that if a precipitate formed after vigorous shaking with
ethyl acetate, it was removed by vacuum filtration through a Büchner funnel.
Figure 10: Partition of acidic layers and ethyl acetate wash before and after removal of precipitate by vacuum filtration
12. pH was determined by using test strips purchased from VWR.
13. Anhydrous
Na2SO4 (99.5%) was purchased from Oakwood and used as received.
14. The disposable filter funnel (60 mL, 10 micron polyethylene frit) was purchased from Chemglass and connected to a 500 mL round-bottomed flask by a 24/40 straight vacuum adaptor.
15. The authors note that if the orange impurity is not sufficiently removed in previous steps, then the concentrated crude mixture appears as a viscous orange tar. However, this impurity is removed by the prescribed recrystallization.
16. Hexanes (≥99%, anhydrous) were purchased from Sigma-Aldrich. The solvent was degassed by purging with argon and passed through alumina columns prior to use with the aid of a solvent purification system.
17. Characterization data for
1:
1H NMR
pdf (500 MHz, CDCl
3) δ 7.67 - 7.58 (m, 2H), 7.37 - 7.27 (m, 5H), 6.70 - 6.59 (m, 2H), 6.27 (s, 1H), 4.62 (d,
J = 5.6 Hz, 2H), 3.97 (s, 2H).
13C NMR
pdf (126 MHz, CDCl
3) δ 167.2, 149.7, 138.7, 128.8 (2C), 128.0, 127.6, 124.0, 114.3, 44.1. FTIR (neat)
ν 3439, 3347, 3236, 1632, 1593, 1499, 1291 cm
-1. The purity was determined to be 98.5% by quantitative NMR
pdf spectroscopy with mesitylene (≥98%) as internal standard. A second reaction performed at the same scale yielded 3.73 g (55%) of the same product, with a purity of 98%.
18. The authors obtained compound
1 as an off-white powder in 98 wt % purity based on qNMR using
1,3,5-trimethoxybenzene as an internal standard. The slight color difference in the final product does not appear meaningful.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The amination of aryl boronic acids provides access to primary anilines which may otherwise be challenging to synthesize. The traditional synthesis of primary anilines involves reduction of nitroarenes. Aromatic nitration can in some cases exhibit poor regioselectivity and functional-group tolerance with electron-deficient aromatics requiring especially harsh nitration conditions.
3 While Pd-catalyzed C-N cross-coupling with an ammonia nucleophile offers a complementary approach, the catalysts can be costly and require special considerations for removal.
4 In recent years, several electrophilic, metal-free methods have been reported to convert aryl boronic acids to anilines.
5,6,7,8 Hydroxylamine-derived reagents, including hydroxylamine-
O-sulfonic acid and
O-(2,4-dinitrophenyl)hydroxylamine, achieve high yields under mild conditions for electron-rich substrates but require elevated temperature and extended reaction times for electron-deficient substrates (Table 1).
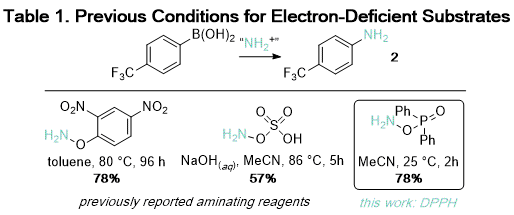
In contrast, amination with DPPH is relatively insensitive to the electronic properties of the substrate. At room temperature, anilines with
para-substituted electron-withdrawing groups including trifluoromethyl (
2), amides (
3), and sulfones (
5) were obtained in good yields (Table 2).
9 DPPH has been used to aminate
nitrogen-containing heterocycles and thioethers, while previously reported hydroxylamine-derived reagents had been found to be incompatible with these motifs.
5,10 Our conditions afforded the desired anilines (
6,
7 and
8) in moderate to good yields (Table 2). One unresolved limitation is the amination of boronic acids
ortho to nitrogen in heterocycles, which is obstructed by rapid protodeborylation.
11
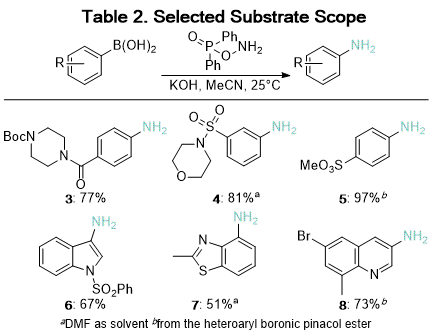
DPPH is a bench-stable, commercially available reagent, and the resulting anilines can be purified without column chromatography.
12 The generality, functional-group tolerance, and mild conditions of this method may be attractive for discovery-scale synthesis or for the late-stage diversification of complex molecules.
Appendix
Chemical Abstracts Nomenclature (Registry Number)
[4-(Benzylcarbamoyl)phenyl]boronic acid: Boronic acid, B-[4-[[(phenylmethyl)amino]carbonyl]phenyl]-; (252663-47-1)
DPPH, O-(Diphenylphosphinyl)hydroxylamine: Phosphinic acid, P,P-diphenyl-, azanyl ester; (72804-96-7)

|
Kasie Leung is an A.B. candidate in Chemistry and Physics at Harvard College. She expects to graduate in 2027 and plans to pursue doctoral studies afterwards. Since joining the Liu Group in 2024, she has worked on redox-active organic molecules for catalysis. |

|
Richard Liu is an assistant professor in the Department of Chemistry and Chemical Biology at Harvard University. He obtained his PhD in 2019 from the Massachusetts Institute of Technology, where he was supervised by Prof. Stephen Buchwald. He worked as a postdoctoral associate with Prof. Timothy Swager, following which he joined Harvard in 2022. |

|
Kanaram Kumawat grew up in Maharashtra, India. He earned his Bachelor of Science degree from Savitribai Phule Pune University and completed his Master's degree at the Indian Institute of Technology (IIT) Guwahati. He is currently a Ph.D. candidate in Organic Chemistry at Indiana University Bloomington, working in the laboratory of Prof. Kevin Brown. His research focuses on the development of novel synthetic methodologies for the preparation of structurally diverse borylated compounds |

|
M. Kevin Brown grew up in the suburbs of Chicago. Kevin received his B.A. degree from Hamilton College in 2002 and then moved to Boston College to pursue graduate studies under the mentorship of Professor Amir Hoveyda. Upon completion of his graduate studies, he began a postdoctoral fellowship in the laboratories of E.J. Corey at Harvard University. In 2011 he began his independent career at Indiana University as an assistant professor and was full professor in 2020. His research interests are focused on the development of new methods for chemical synthesis. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved