Org. Synth. 2005, 81, 42
DOI: 10.15227/orgsyn.081.0042
PALLADIUM CATALYZED CROSS-COUPLING OF (Z)-1-HEPTENYLDIMETHYLSILANOL WITH 4-IODOANISOLE: (Z)-1-HEPTENYL)-4-METHOXYBENZENE
[Benzene,1-(1Z)-1-heptenyl-4-methoxy-]
Submitted by Scott E. Denmark and Zhigang Wang
1.
Checked by Matthew Campbell and Dennis P. Curran.
1. Procedure
A.
1-Iodo-1-heptyne.
2 A solution of
9.62 g (100 mmol) of 1-heptyne (Note 1) in
50 mL of hexane (Note 2) is placed in a
flame-dried, three-necked, 500-mL, round-bottomed flask equipped with a large
stirbar, a
pressure equalizing addition funnel, and an
N2 inlet. The solution is cooled to −50 to −55°C
(Note 3) and a solution of
n-BuLi in
hexane (61.0 mL, 1.65M, 100 mmol) (Note 4) is then added dropwise over 30 min. The resulting thick suspension is stirred at −70°C for 1.5 hr. A solution of
25.4 g (100 mmol) of Iodine (Note 5) in
130 mL of diethyl ether (Note 6) is added over 45 min. The cooling bath is removed and the reaction solution is allowed to warm to room temperature.
Water (100 mL) is added and the resulting mixture is stirred at room temperature for 10 min. The aqueous layer is separated and extracted with
100 mL of hexane, and the combined organic phases are washed with two
200-mL portions of water,
50 mL of 20% aqueous sodium thiosulfate (Na2S2O3) solution, and then dried over
anhydrous MgSO4 (Note 7). The solvents are removed by rotary evaporation
(Note 8) and the residue is dissolved in
30 mL of pentane (Note 9) and the solution is then passed through a short column of
silica gel
(Note 10) followed by further elution with
250 mL of pentane. The solvent is removed by rotary evaporation and the residue is purified by Kugelrohr distillation to afford
17.9-18.5 g (
81-83%) of
1-iodo-1-heptyne as a colorless liquid,
bp 60-65°C at 1.2 mm (lit.
2 55-60°C at 5 mm) (Notes
11,
12).
B.
(Z)-1-Iodo-1-heptene.2 A solution of
8.52 g of (112 mmol) of borane-dimethylsulfide complex (Note 13) in
100 mL of ether is added to a
flame-dried, three-necked, 300-mL, round-bottomed flask equipped with
stirbar,
temperature probe, and
N2 inlet. The solution is cooled to 5°C with an
ice-bath.
Cyclohexene (18.4 g, 224 mmol) (Note 14) is then added by
syringe over 10 min while keeping the temperature below 15°C. The mixture is stirred at 5°C for 15 min. A white solid precipitates either towards the end of the addition or during the subsequent stirring period. The reaction mixture is allowed to warm to room temperature and is stirred for 1 hr. The non-homogeneous solution is cooled to 2-3°C and
22.7 g (102 mmol) of 1-iodo-1-heptyne is added by
syringe over 10 min. The reaction mixture is stirred at 2-3°C for 30 min, and then the cooling bath is removed and the mixture is stirred at room temperature for 1 hr. The solution is cooled to 2-3°C and
51 mL of acetic acid (AcOH) (Note 15) is slowly added. The ice-bath is removed after the addition of AcOH is completed and the mixture is stirred at room temperature for 2 hr.
Ether (100 mL) is added and the solution is washed with
four 75-mL portions of water, dried over
anhydrous MgSO4, filtered, and concentrated by rotary evaporation to give a pale yellow liquid which is dissolved in
50 mL of pentane. The solution is passed through a short column of
silica gel
(Note 10) followed by further elution with
250 mL of pentane. The solvent is removed by rotary evaporation to give the crude product which is purified by Kugelrohr distillation to afford
15.7 g (
69%) of
(Z)-1-iodo-1-heptene as a colorless liquid,
bp 75-80°C at 1.5 mm (lit.
2 60-63°C at 5.5 mm) (Notes
11,
16).
C.
(Z)-1-(1-Heptenyl)-1,1-dimethylsilanol. A solution of
13.0 g (58 mmol) of (Z)-1-iodo-1-heptene in
50 mL of ether is placed in a
flame-dried, three-necked, 300-mL, round-bottomed flask equipped with stirbar,
septum,
temperature probe, and
N2 inlet. The solution is cooled to −72°C and a solution of
n-BuLi in
hexane (Note 4) (35.6 mL, 1.63M, 58 mmol) is then added by
syringe over 15 min. The mixture is stirred at −72°C for 30 min and then a solution of
4.30 g (19.3 mmol) of hexamethylcyclotrisiloxane (Note 17) in
50 mL of ether is added over 15 min. The cooling bath is removed, and the reaction solution is allowed to warm to room temperature and stirred for 14 hr. The solution is then cooled to 0°C and
50 mL of water is slowly added to quench the reaction. The aqueous layer is separated and extracted with
three 50-mL portions of diethyl ether. The combined organic phases are washed with
30 mL of water and
two 30-mL portions of brine, dried over
anhydrous MgSO4, filtered, and concentrated by
rotary evaporator and vacuum drying to give a crude product which is distilled to afford
7.34 g (
73%) of
(Z)-1-(1-heptenyl)-1,1-dimethylsilanol as a colorless liquid,
bp 54-55°C at 0.15 mm (lit.
3 120°C at 0.9 mm) (Notes
18,
19).
D.
(Z)-1-(1-Heptenyl)-4-methoxybenzene. A solution of
4.14 g (24 mmol) of (Z)-1-(1-heptenyl)-1,1-dimethylsilanol in
2.0 mL of THF is added to a
flame-dried, 250 mL, three-necked, round-bottomed flask equipped with a stirbar,
addition funnel,
temperature probe, and
argon inlet. A solution of
tetrabutylammonium fluoride in THF (40 mL, 1.0M, 40 mmol) (Note 20) is then added over 5 min. The mixture is stirred at room temperature for 10 min and then
0.575 g (1.0 mmol) of Pd(dba)2 (Note 21) is added. A solution of
4-iodoanisole in THF (20 mL, 1.0M, 20 mmol) (Note 22) is added slowly through the
addition funnel (Note 23) such that the temperature of the solution is kept between 30-33°C (addition time 30 min). The mixture is stirred for 30 min after complete addition of
4-iodoanisole and then
70 mL of diethyl ether is added and the mixture is stirred for an additional 10 min. The mixture is passed through a
short column of silica gel (Note 10) followed by further elution with
200 mL of diethyl ether. The combined eluate is concentrated by rotary evaporation and vacuum drying to give a residue which is purified by Kugelrohr distillation to afford a colorless or light yellow liquid. This liquid is further purified by chromatography
(Note 24) followed by Kugelrohr distillation to afford
3.29 g (
80%) of
(Z)-1-(1-heptenyl)-4-methoxybenzene as a colorless oil, bp 90-95°C at 0.1 mm (lit.3 80-85°C at 1.3 mm) (Notes
11,
25).
2. Notes
1.
1-Heptyne was purchased from GFS Chemicals Inc. and was used without further purification.
2.
Hexane was purchased from Fisher Scientific Company and was freshly distilled from
sodium.
3.
Unless otherwise noted, all temperatures refer to internal temperatures measured with Teflon-coated thermocouples.
4.
Butyllithium was purchased from FMC Corporation Lithium Division and its concentration was determined by double titration by the Gilman method.
45.
I2 was purchased from Mallinckrodt Inc. and was used as received.
6.
Ether was freshly distilled from
sodium/
benzophenone.
7.
Anhydrous MgSO4 was purchased from Fisher Scientific Company.
8.
The vacuum for the rotary evaporator is about 15 mm.
9.
Pentane was purchased from Fisher Scientific Company and was distilled from CaCl2.
10.
Silica gel was purchased from VWR Scientific (230-400 mesh). A 45-mm diameter column was employed and
50 g silica gel was loaded as a
pentane slurry.
11.
Boiling points (bp) correspond to uncorrected air-bath temperatures in the Buchi GKR-50 Kugelrohr.
12.
The analytical data are as follows:
1H NMR (500 MHz, CDCl
3) δ: 0.90 (t,
J = 7.1, 3 H), 1.34 (m, 4 H), 1.51 (m, 2 H), 2.35 (t,
J = 7.1, 2 H);
13C NMR (125.6 MHz, CDCl
3) δ: −7.2, 13.9, 20.7, 22.1, 28.2, 30.9, 94.6; IR (NaCl) cm
−1: 2956, 2932, 2859, 1462; Anal. Calcd. for C
7H
11I: C, 37.86; H, 4.99; I, 57.15; Found: C, 37.90; H, 5.08; I, 57.50.
13.
Borane-methyl sulfide complex (neat) was purchased from Aldrich Chemical Company, Inc. and was used as received.
14.
Cyclohexene was purchased from Aldrich Chemical Company, Inc. and was used as received.
15.
Acetic acid (100%) was purchased from J. T. Baker Company and was used as received.
16.
The checkers obtained the product in
65-74% yield. The analytical data are as follows:
1H NMR
pdf (500 MHz, CDCl
3) δ: 0.90 (t, J = 7.1, 3 H), 1.32 (m, 4 H), 1.34 (m, 2 H), 2.13 (m, 2 H), 6.17 (m, 2 H);
13C NMR (125.6 MHz, CDCl
3) δ: 14.0, 22.5, 27.6, 31.3, 34.6, 82.1, 141.5; IR (NaCl) cm
−1: 2956, 2927, 2857, 1608, 1463, 1276; GC analysis:
tR (
Z) 6.34 min (99.3%),
tR (
E) 6.57 min (0.7%) (HP-5, 150 °C, 15 psi); Anal. Calcd. for C
7H
13I: C, 37.52; H, 5.85; I, 56.63; Found: C, 37.72; H, 5.96; I, 56.81.
17.
Hexamethylcyclotrisiloxane was purchased from Gelest Inc. and was used as received.
18.
The
(Z)-1,1-dimethyl-1-heptenylsilanol easily forms
1,1,1',1'-tetramethyl-1,1'-diheptenyldisiloxane. The crude product was distilled (60-62°C at 0.5 mm) to afford
8.02 g (
80%) of (
Z)-1,1-dimethyl-1-heptenylsilanol containing
2-5% of
1,1,1',1'-tetramethyl-1,1'-diheptenyldisiloxane. To obtain analytically pure product, the silanol was distilled twice again
(bp 54-55°C, 0.15 mm).
19.
The checkers obtained the product in
67-89% yield. The analytical data are as follows:
1H NMR
pdf (500 MHz, CDCl
3) δ: 0.24 (s, 6 H), 0.89 (t, J = 7.1, 3 H), 1.30 (m, 4 H), 1.39 (m, 2 H), 1.69 (br, 1 H), 2.19 (m, 2 H), 5.47 (dt, J = 14.1, 1.3, 1 H), 6.36 (dt, J = 14.1, 7.5, 1 H);
13C NMR (125.6 MHz, CDCl
3) δ: 1.7, 14.0, 22.6, 29.3, 31.5, 33.6, 127.8, 151.6; IR (NaCl) cm
−1: 3274 (br), 2959, 2928, 2858, 1607, 1465, 1253, 1067, 864, 785; GC analysis:
tR 7.15 min (HP-1, 100 °C, 10 psi); Anal. Calcd. for C
9H
20OSi: C, 62.72; H, 11.70; Found: C, 62.56; H, 12.04.
20.
A solution of
tetrabutylammonium fluoride (TBAF) in THF (1.0M) is prepared from colorless, crystalline tetrabutylammonium fluoride trihydrate purchased from Fluka Chemical Corp.21.
Pd(dba)2 was prepared by the literature procedure without recrystallization from CHCl3;
5 See
(Note 8),
Org. Synth.,
2004,
81, 57.
22.
4-Iodoanisole was purchased from Aldrich Chemical Company, Inc., and was purified by chromatography on silica gel (
hexane/EtOAc, 50/1) prior to use.
23.
The coupling reaction is very exothermic. The reaction temperature can be controlled by the rate of addition of
4-iodoanisole.
24.
A 55 mm diameter column is employed and
200 g silica gel is used (
pentane/EtOAc, 50/1).
25.
The checkers obtained the product in
90-97% yield. The analytical data are as follows:
1H NMR
pdf (400 MHz, CDCl
3) δ: 0.89 (t, J = 7.1, 3 H), 1.31 (m, 4 H), 1.45 (qn, J = 7.3, 2 H), 2.31 (qd, J = 7.3, 2 H), 3.81 (s, 3 H), 5.57 (dt, J = 11.7, 7.1, 1 H), 6.33 (d, J = 11.7, 1 H), 6.87 (d, J = 8.8, 2 H), 7.22 (d, J = 8.8, 2 H);
13C NMR (100.6 MHz, CDCl
3) δ: 13.9, 22.6, 28.6, 29.7, 31.6, 55.0, 113.5, 128.0, 129.9, 130.5, 131.5, 158.1; IR (NaCl) cm
−1: 3005, 2956, 2927, 2856, 1608, 1511, 1463, 1301, 1249, 1175, 1038, 837; GC analysis:
tR(
Z) 6.05 min (
99.1%),
tR(
E) 6.53 min (
0.9%) (HP-5, 250 °C, 15psi); Anal. Calcd. for C
14H
20O: C, 82.30; H, 9.87; Found: C, 82.25; H, 9.96.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Palladium-catalyzed, cross-coupling reactions of organosilicon reagents with aryl heteroaryl and alkenyl halides have emerged in recent years as a synthetically viable alternative to the traditional organotin (Stille) and organoboron (Suzuki) cross-coupling reactions. The most extensively investigated of these processes involves the use of mono-, di-, and trifluorosilanes as the coupling partners activated by a fluoride source.
6 Recently, aryl trialkoxysiliconates have also shown promise for carbon-carbon bond formation under similar conditions.
7Our recent demonstration that a silicon-oxygen bond is the key structural feature that lends high reactivity of organosilicon compounds toward cross-coupling has led to the development of a wide range of silicon-based substrates for this process.
3,8 The most thoroughly investigated of these derivatives are the dialkylsilanols. These compounds are easily prepared by a number of different methods, the most common of which illustrated here is the reaction of organolithium compounds with
hexamethylcyclotrisiloxane (D
3).
9,10 The addition is general for alkenyl-, aryl-, heteroaryl- and alkynyllithium agents and all three silicon groups in D
3 are available.
The palladium-catalyzed cross-coupling of alkenylsilanols has been extensively studied with respect to the structure of both the silicon component and the acceptor halide. The preferred catalyst for coupling of aryl iodides is Pd(dba)2 and for aryl bromides it is [allylPdCl]2. The most effective promoter is tetrabutylammonium fluoride used as a 1.0M solution in THF. In general the coupling reactions occur under mild conditions (room temperature, in 10 min to 12 hr) and some are even exothermic.
Varying substitution patterns have been studied and while the rate of the coupling is attenuated by steric crowding of the dimethylsilanol, the yields and stereospecificities are still high. In all cases studied to date, the coupling reactions proceed with retention of configuration. The examples compiled in Figure 1 represent successful couplings of a number of classes of alkenylsilanols in combination with both aryl and alkenyl halides.
8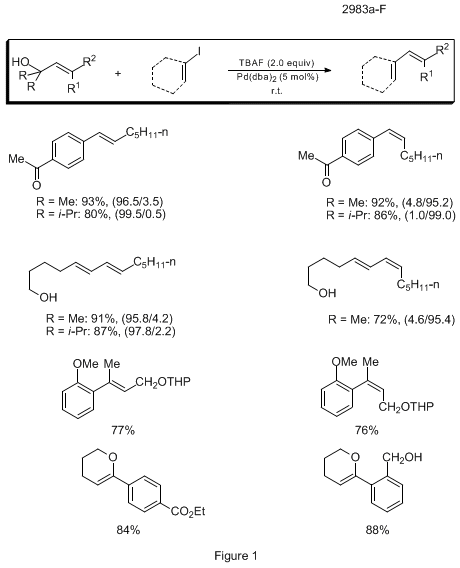
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
1-Heptyne:
1-Heptyne; (628-71-7)
Cyclohexene; (110-83-8)
1-Iodo-1-heptyne:
1-Heptyne, 1-iodo-; (54573-13-6)
Butyllithium:
Lithium, butyl; (109-72-8)
(Z)-1-Iodo-1-Heptene:
1-Heptene, 1-iodo-, (1Z)-; (63318-29-6)
Borane-dimethylsulfide complex:
Boron, trihydro[thiobis[methane]]-(T-4)-; (13292-87-0)
(Z)-1,1-Dimethyl-1-heptenylsilanol:
Silanol, (1Z)-1-heptenyldimethyl-; (261717-40-2)
(Z)-1-Heptenyl-4-methoxybenzene:
Benzene, 1-(1Z)-1-heptenyl-4-methoxy-; (80638-85-3)
Iodine; (7553-56-2)
4-Iodoanisole:
Benzene, 1-iodo-4-methoxy-; (696-62-8)
Hexamethylcyclotrisiloxane:
Cyclotrisiloxane, hexamethyl-; (54-05-9)
Tetrabutylammonium fluoride trihydrate:
1-Butanaminium, N, N, N-tributyl, fluoride, trihydrate; (87749-50-6)
Pd(dba)2: Palladium, bis[(1,2,4,5-()-1,5-diphenyl-1,4-pentadien-3-one]-; (32005-36-0)
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved