Org. Synth. 2011, 88, 102-108
DOI: 10.15227/orgsyn.088.0102
Discussion Addendum for: PALLADIUM CATALYZED CROSS-COUPLING OF (Z)-1-HEPTENYLDIMETHYLSILANOL WITH 4-IODOANISOLE: (Z)-(1-HEPTENYL)-4-METHOXYBENZENE
Submitted by Scott E. Denmark
* and Jack Hung-Chang Liu
1.
Discussion
The cross-coupling of organosilanols has emerged as a viable alternative to the classical methods of Suzuki (boronic acids), Stille (stannanes) and Negishi (organozincs).
2 The major developments over the past years have been the significant expansion of the scope of the organosilanol (or silanol precursor) and the introduction of non-fluoride activation of the silanols. Both advances will be summarized here.
Scope of Organosilanol Donor
Over the past ten years, a wide range of organosilanols have been prepared
3 and shown to be competent partners in the fluoride-activated cross coupling with aromatic and olefinic halides (including bromides
4) and triflates.
5 Most notable has been the extension to pyranylsilanols,
6 cyclic siloxanes generated by intramolecular hydrosilylation,
7 intramolecular silylformylation,
8 and ring-closing metathesis
9 (Figure 1). The latter tandem process (RCM-cross-coupling) was featured in a total synthesis of (+)-brasilenyne (Figure 2).
10 In addition, silylcarbocyclization-formylation
11 allowed for the construction of the pyrrolidine core of isodomoic acids G and H (Figure 3).
12 The final cross-coupling step involved a fluoride mediated process that employed a buffered form of TBAF (octahydrate).
Figure 1. Newer variations of fluoride-promoted, silicon-based-cross coupling reactions.
Figure 2. Fluoride-promoted intramolecular alkenyl-alkenyl cross-coupling for the syntheses of brasilenyne.
Figure 3. Fluoride-promoted intermolecular alkenyl-alkenyl cross-coupling for the syntheses of isodomoic acids G and H.
Fluoride-Free Cross-Coupling Reactions
By far the most important advance in the past five years has been the discovery of a preparatively useful and mechanistically distinct
13 pathway for cross-coupling of organosilanols that employs various Brønsted bases as activators.
2d This discovery has allowed for a wider range of coupling partners to be incorporated and also for milder reaction conditions to be employed. For example is it now possible to effect the cross-coupling of simple alkenylsilanols and alkynylsilanols (with KOTMS),
14 arylsilanols (with Cs
2CO
3),
15 and 2-indolylsilanols (with KO
t-Bu),
16 and isoxazolinylsilanols (with KO
t-Bu)
17 (Figure 4).
Figure 4. Variations of fluoride-free, cross-coupling reactions.
The ability to couple silanols under fluoride-free conditions has allowed the introduction of the preformed silanolate salts as viable coupling partners. The salts can be easily prepared by deprotonation with NaH or KH and are, in general, stable, free flowing powders. The silanolates couple directly without the need for added bases or activators. Accordingly, heterarylsilanolates derived from indoles, thiophenes, furans
18 as well as a wide range of aromatic silanolates
19 have been successfully employed. Finally, alkenylsilanolates also undergo high yielding and highly stereospecific cross-coupling with aryl chlorides
20 (Figure 5).
Figure 5. Cross-coupling reactions of preformed silanolate salts.
The demonstration of both fluoride and non-fluoride activation for silicon-based cross-coupling has led to the development of a conjunctive reagent that allows for sequential coupling at separate ends of a 1,4-butadiene unit.
21 This application was featured in the total synthesis of RK-397 (Figure 6).
22Figure 6. Sequential cross-coupling of a 1,4-bissilyl-1,3-butadiene.
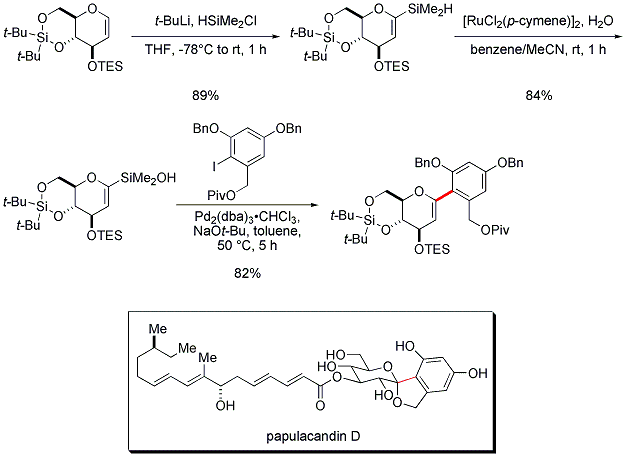
The construction of the key aryl glycosidic bond en route to papulacandin D highlights the synthetic utility of the Brønsted base activation method (Figure 7).
23 The glycal silanol would not withstand activation by fluoride and the resorcinol coupling partner is highly deactivated. Nevertheless, the desired coupling could be achieved by the action of sodium
tert-butoxide at 50 °C in the presence of Pd
2(dba)
3•CHCl
3. The coupled product contains the entire carbon framework of the sugar fragment of papulacandin D.
Figure 7. Fluoride-free alkenyl-aryl cross-coupling for the total synthesis of papulacandin D.
 |
Scott E. Denmark was born in Lynbrook, New York on 17 June 1953. He obtained an S.B. degree from MIT in 1975 and his D.Sc.Tech. (with Albert Eschenmoser) from the ETH Zürich in 1980. That same year he began his career at the University of Illinois. He was promoted to associate professor in 1986, to full professor in 1987 and since 1991 he has been the Reynold C. Fuson Professor of Chemistry. His research interests include the invention of new synthetic reactions, exploratory organoelement chemistry and the origin of stereocontrol in fundamental carbon-carbon bond forming processes. |
 |
Jack Hung-Chang Liu was born in Taipei, Taiwan in 1979. He obtained a Hon. B. Sc. Degree at University of Toronto in 2002 (Working with Robert A. Batey and Mark Lautens). He then joined the research group of Scott E. Denmark at University of Illinois at Urbana-Champaign, focusing on the development and application of silicon-based cross-coupling. After obtaining his Ph. D. degree in 2009, he moved to University of California, Berkeley for post-doctoral research under F. Dean Toste. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved