Org. Synth. 2006, 83, 97
DOI: 10.15227/orgsyn.083.0097
SYNTHESIS OF (−)-(S,S)- BIS(4-ISOPROPYLOXAZOLINE)
[(4S, 4S')-2,2'-(Propane-2,2-diyl)bis(4-isopropyl-4,5-dihydrooxazole)]
Submitted by David A. Evans
1, Keith A. Woerpel
1,2, Bernd Nosse
3, Andreas Schall
3, Yogesh Shinde
3, Eva Jezek
3, Mohammad Mahbubul Haque
3, R. B. Chhor
3, and Oliver Reiser
3.
Checked by Peter Wipf and Nilukshi Jayasuriya
4.
1. Procedure
A. (−)-(S,S)-N,N'-Bis(1-hydroxymethyl-2-methylpropyl)-2,2-dimethyl-malonamide (3): An oven-dried 250 mL, 3-necked round-bottom flask equipped with a stirring bar and two 50 mL pressure-equalizing addition funnels connected to a mineral oil bubbler is purged with nitrogen and charged with (L)-valinol (2, 5.13 g, 0.050 mol, Note 1). The flask is immersed in an ice bath at 0 °C and triethylamine (17.4 mL, 0.124 mol, Note 2) is added dropwise via the first addition funnel. 2,2-Dimethylpropanedioyl dichloride (3.3 mL, 0.25 mol, Note 3) in dry dichloromethane (25 mL, Note 4) is then added dropwise over 25 minutes via the second addition funnel. The internal temperature increases from 0 °C to 10 °C during the addition. Subsequently, the ice bath is removed and the reaction mixture is allowed to warm to room temperature. Stirring is continued for 45 min, resulting in a colorless precipitate that is dissolved by addition of dry dichloromethane (120 mL). After addition of aqueous HCl (1 N, 30 mL), the aqueous layer is separated and extracted with dichloromethane (3 × 15 mL). The combined organic layers are washed with saturated NaHCO3 solution (30 mL) and brine (30 mL), dried over MgSO4, filtered and concentrated in vacuo to afford crude 3 as a pale yellow solid. Recrystallization of the crude product from ethyl acetate (40 mL) yields 3 (4.30 g, 14.2 mmol, 57%) as white crystals. The mother liquor is concentrated and the residue recrystallized from ethyl acetate (10 mL) to yield a second crop of 3 (1.60 g, 5.27 mmol, 21%); the process is repeated to yield a third crop of 3 (0.440 g, 1.45 mmol, 6%, total yield: 6.40 g, 21.1 mmol, 84%, Note 5).
B. (−)-(S,S)-Bis(4-isopropyloxazoline) (4): An oven-dried 500 mL, 2-necked round-bottom flask equipped with a stirring bar and a 50 mL, pressure-equalizing addition funnel connected to a mineral oil bubbler is purged with nitrogen and charged with (−)-(S,S)-N,N'-bis-(1-hydroxymethyl-2-methylpropyl)-2,2-dimethylmalonamide (3, 5.5 g, 18.4 mmol), 4-dimethylaminopyridine (0.204 g, 1.67 mmol, Note 6) and dry dichloromethane (130 mL, Note 4). The flask is immersed in a water bath at room temperature and triethylamine (10.25 mL, 73.4 mmol, Note 2) is added slowly via syringe. Subsequently, tosyl chloride (7.10 g, 37 mmol, 2.0 equiv., Note 7), dissolved in dry dichloromethane (15 mL), is added dropwise over 30 minutes via the addition funnel. After completion of the addition, the funnel is rinsed with dry dichloromethane (2.5 mL) and the reaction mixture is stirred for an additional 27 h at room temperature (Note 8). The reaction mixture is treated with saturated NH4Cl solution (70 mL) followed by water (40 mL). The aqueous layer is separated and extracted with dichloromethane (3 × 55 mL), and the combined organic layers are dried over MgSO4. The organic solution is filtered and concentrated under vacuum. The oily residue is treated with hot pentane (40 mL, Note 9), stirred for 5 min and the supernatant liquid is decanted. This procedure is repeated three times and the collected pentane layers are combined and concentrated under vacuum to yield 4 (4.05 g, 15.2 mmol, 83%, Note 10) as a colorless oil.
2. Notes
1.
(L)-Valinol was prepared from
(L)-valine in
81% yield in an analogous way to the reduction of
(S)-tert-leucine to
(S)-tert-leucinol according to Evans, D. A.; Peterson, G. S.; Johnson, J. S.; Barnes, D. M.; Campos, K. R.; Woerpel, K. A.
J. Org. Chem. 1998,
63, 4541, or purchased from Aldrich Chemical Company, Inc.
2.
Triethylamine was obtained from Alfa Aesar and was distilled prior to use from
calcium hydride under nitrogen.
3.
2,2-Dimethylpropanedioyl dichloride was purchased from Aldrich Chemical Company, Inc. or synthesized from
2,2-dimethylmalonic acid in
90% yield according to Evans, D. A.; Peterson, G. S.; Johnson, J. S.; Barnes, D. M.; Campos, K. R.; Woerpel, K. A.
J. Org. Chem. 1998,
63, 4541.
4.
Dichloromethane was purified by filtration through activated
alumina.
5.
Physical properties and spectral data for
3 are as follows: R
f 0.25 (
EtOAc:MeOH, 95:5);
[α]D24 −6.0 (c 0.50, CH2Cl2); mp
98–99 °C;
1H NMR
pdf (300 MHz, CDCl
3) δ: 0.92 (d,
J = 6.8 Hz, 6 H), 0.96 (d,
J = 6.8 Hz, 6 H), 1.50 (s, 6 H), 1.82 (oct,
J = 6.8 Hz, 2 H), 2.66 (bs, 2 H), 3.52 (m, 2 H), 3.69–3.86 (m, 4 H), 6.41 (d,
J = 8.6 Hz, 2 H);
13C NMR
pdf (75 MHz, CDCl
3) δ: 18.8, 19.6, 23.7, 29.1, 50.2, 57.1, 63.5, 174.5; IR (KBr) cm
−1 3349, 3380, 2962, 2886, 1658, 1530, 1049, 1033; MS (ES)
m/z (%) 405.2 (20), 325.2 ([M + Na]
+, 100), 285.2 (10); HRMS (ES) calcd. for C
15H
30N
2O
4Na 325.2103, found 325.2084; Anal. calcd. for C
15H
30N
2O
4: C, 59.57; H, 10.00; N, 9.26. found: C, 59.01; H, 10.12; N, 9.11.
6.
4-Dimethylaminopyridine was obtained from the Aldrich Chemical Company, Inc and used as received.
7.
Tosyl chloride was obtained from the Aldrich Chemical Company, Inc and used as received.
8.
After the reaction mixture was stirred for 27 h, the submitters noticed crystalline solid precipitate. The solid precipitate was dissolved by adding
dichloromethane (50 mL) prior to workup. The checkers, however, did not observe any precipitate.
9.
In some experiments, a cloudy precipitate was formed, which was then dissolved in
dichloromethane (2.5 mL per mmol of 3) prior to workup.
10.
An analytically pure sample for characterization purposes was obtained by Kugelrohr distillation (95-100 °C, 0.5 mmHg) of the crude material. Physical properties and spectral data for
4 are as follows R
f 0.25 (
CH2Cl2/MeOH, 19:1);
[α]D24 −107.5 (c 1.0, CH2Cl2);
1H NMR
pdf (300 MHz, CDCl
3) δ: 0.85 (d,
J = 6.8 Hz, 6 H), 0.91 (d,
J = 6.8 Hz, 6 H), 1.51 (s, 6 H), 1.88–1.73 (m, 2 H), 4.06–3.93 (m, 4 H), 4.26–4.15 (m, 2 H);
13C NMR
pdf (75 MHz, CDCl
3) δ: 17.3, 18.5, 24.4, 32.2, 38.5, 69.9, 71.5, 168.7; IR (film) cm
−1 3308, 2960, 2874, 1746, 1525, 1353, 1303, 1037, 1017, 895, 815, 714; MS (EI)
m/z (%) 266 (M
+, 15), 265 (30), 223 (100), 195 (30), 155 (65), 137 (97), 110 (50); HRMS (EI) calcd for C
15H
26N
2O
2 266.1994, found 266.1987; Anal. calcd for C
15H
26N
2O
2: C, 67.63; H, 9.84; N, 10.52. found C, 66.65; H, 9.81; N, 10.08.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Since the discovery of oxazolines as ligands for catalysis,
5 bis(oxazolines)
6 have proved to be privileged structures because they promote a great number of metal-catalyzed transformations with unprecedented selectivity. Most commonly, bis(oxazolines) can be obtained from amino alcohols, either by a two-step condensation/cyclization sequence with acid chlorides as described here, or by condensation with dinitriles in the presence of a metal salt such as
zinc(II) chloride7,
trifluoro-methanesulfonic acid8 or stepwise formation of the corresponding imidates
9 followed by cyclization. While the commercially available
tert-butyl-substituted bis(oxazoline)
510,11 often gives rise to the highest selectivities, a number of applications
12 have been developed in which the
bis(4-isopropyloxazoline) 4 or the phenyl-substituted bis(oxazoline)
6 will give similar or even better results. Moreover, for the synthesis of
5 the unnatural and therefore expensive amino acid
tert-leucine is required. Consequently,
bis(4-isopropyloxazoline) 4, available as either enantiomer from inexpensive (
S)- or
(R)-valine, is an attractive alternative for large scale applications if equally good enantioselectivity can be achieved.
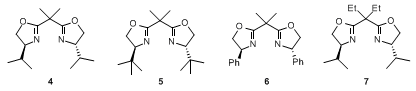
The procedure described here for
bis(4-isopropyloxazoline) 4 closely resembles the previously reported protocols for the
bis(4-ethyloxazoline) 713 and the bis(4-
tert-butyloxazoline) ligand
5.
10 The main differences can be found in the workup conditions, which accommodate the quite different solubility and crystallization properties of intermediates and final ligands.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
2,2-Dimethyl-propanedioyl dichloride:
Propanedioyl dichloride, dimethyl-; 5659-93-8
(L)-Valinol:
1-Butanol, 2-amino-3-methyl-, (2S)-; 2026-48-4
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved