Org. Synth. 2007, 84, 88
DOI: 10.15227/orgsyn.084.0088
SYNTHESIS OF YNAMIDES BY N-ALKYNYLATION OF AMINE DERIVATIVES. PREPARATION OF N-ALLYL-N-(METHOXYCARBONYL)-1,3-DECADIYNYLAMINE
Submitted by Amanda L. Kohnen, Joshua R. Dunetz, and Rick L. Danheiser
1.
Checked by Scott E. Denmark and Xiaorong Liu.
1. Procedure
A. N-(Methoxycarbonyl)-2-propenylamine (2) (Note 1). A 250-mL, three-necked, round-bottomed flask equipped with a magnetic stir bar, an argon gas inlet tube, a rubber septum, which is pierced with a Teflon-coated thermocouple, and a 200-mL pressure-equalizing addition funnel fitted with a rubber septum (Note 2) is charged via syringe through the septum with allylamine (1) (14.0 mL, 10.68 g, 187 mmol, 2.1 equiv) (Note 3) and 100 mL of dichloromethane (Note 4). The solution is cooled to 0 °C (internal temperature) in an ice-water bath and a solution of methyl chloroformate (7.0 mL, 8.6 g, 91 mmol) (Note 5) in 30 mL of dichloromethane is added dropwise via the addition funnel over 10 min. The resulting white mixture is allowed to warm to room temperature over 30 min and then transferred to a 500-mL separatory funnel and extracted with 50 mL of aqueous 1M HCl solution, 50 mL of saturated aqueous NaHCO3 solution, and 50 mL of saturated aqueous NaCl solution. The organic phase is dried over MgSO4, filtered, and concentrated by rotary evaporation (23 °C, 16 mmHg) to afford 10.12–10.26 g (97–98%) of carbamate 2 as a colorless oil (Note 6).
B. 1-Bromo-1,3-decadiyne (4). Into a 200-mL, round-bottomed flask equipped with a magnetic stir bar (Note 2) is added 1,3-decadiyne (3) (5.66 g, 42.2 mmol) and 100 mL of acetone (Note 7). The flask is equipped with a rubber septum and argon inlet needle. N-Bromosuccinimide (8.26 g, 46.4 mmol, 1.1 equiv) (Note 8) and AgNO3 (0.717 g, 4.22 mmol, 0.10 equiv) (Note 9) are each added in one portion through temporary removal of the septum. The rubber septum is replaced with an argon inlet adapter, the flask is wrapped with aluminum foil (Note 10), and the reaction mixture is stirred at room temperature for 22 h (Note 11). The resulting cloudy orange mixture is transferred to a 500-mL separatory funnel with the aid of 100 mL of pentanes and washed with two 50-mL portions of saturated aqueous Na2S2O3 solution (Note 12). The combined aqueous layers are extracted with two 50-mL portions of pentanes and the combined organic phases are washed with 100 mL of saturated NaCl solution, dried over MgSO4, filtered, and concentrated by rotary evaporation (23 °C, 16 mmHg) to provide 8.18–8.25 g (91–92%) of bromodiyne 4 as a red oil (Notes 13, 14).
C. N-(Methoxycarbonyl)-N-(2-propenyl)-1,3-decadiynylamide (5). Into a 500-mL, three-necked, round-bottomed flask is weighed carbamate 2 (3.98 g, 34.6 mmol). The flask is equipped with a large magnetic stir bar, an argon inlet adapter, rubber septum, and a 125-mL pressure-equalizing addition funnel fitted with a rubber septum (Note 1). Tetrahydrofuran (130 mL) (Note 15) and pyridine (70 mL, 68.0 g, 867 mmol, 25 equiv) (Note 16) are added via syringe through the septum. The solution is cooled to 0 °C (internal temperature) in an ice-water bath and 38 mL of potassium hexamethyldisilazide solution (0.91 M in THF, 34.6 mmol, 1.0 equiv) (Note 17) is added via syringe over 10 min. The peach-colored slurry is allowed to warm to room temperature over 15 min and then copper(I) iodide (6.58 g, 34.6 mmol, 1.0 equiv) (Notes 18, 19) is added in one portion by temporarily removing the septum. The resulting yellow-green mixture is stirred at room temperature for 2 h. A solution of bromodiyne 4 (8.47 g, 39.8 mmol, 1.15 equiv) in 40 mL of THF is then added via the addition funnel over 45 min (5 mL THF rinse) (Note 20). The flask and addition funnel are wrapped with aluminum foil to protect the bromodiyne 4 from light and the resulting mixture is stirred at room temperature for 20 h (Note 21). The dark red-brown reaction mixture is transferred to a 1-L separatory funnel, diluted with 150 mL of diethyl ether, and washed with four 100-mL portions of a 3:1 mixture of saturated NaCl solution and concentrated NH4OH solution (Note 22). The blue aqueous layers are extracted with two 100-mL portions of Et2O, and the combined organic layers are washed with two 200-mL portions of 1M HCl solution, 200 mL of saturated NaCl solution, dried over MgSO4, filtered, and concentrated by rotary evaporation (40 °C, 16 mmHg) to afford 18.5 g of dark red oil. This material is transferred to a column (12 × 8 cm) of 300 g of silica gel and eluted with 500 mL of hexanes. Elution is then continued with 5% EtOAc-hexanes and 50-mL fractions are collected. Fractions 14 to 51 are combined and concentrated by rotary evaporation (23 °C, 16 mmHg) to provide 6.88 g (80%) of diynamide 5 as a red oil (Notes 23, 24).
2. Notes
1.
t-Butyl and ethyl
N-allylcarbamate are commercially available. The method described here for the synthesis of the methyl derivative
2 illustrates a general procedure for the preparation of carbamates which usually delivers the desired compounds in near-quantitative yield and in a state of high purity, suitable for use in the
N-alkynylation reaction without further purification.
2.
The apparatus was flame-dried under reduced pressure (0.08 mmHg) and then maintained under an atmosphere of argon during the course of the reaction.
3.
Allylamine (98%) was purchased from Alfa Aesar and used without purification.
4.
Dichloromethane was purified by pressure filtration under argon through activated alumina.
5.
Methyl chloroformate (99%) was obtained from Aldrich Chemical Company, Inc. and used as received.
6.
The product exhibits the following spectroscopic properties: R
f = 0.17 (hexanes/EtOAc, 4/1); IR (neat) cm
−1: 3337, 3084, 2952, 1702, 1534;
1H NMR
pdf (400 MHz, CDCl
3) δ: 3.68 (s, 3 H), 3.81 (app s, 2 H), 4.73 (br s, 1 H), 5.13 (dd,
J = 10.2, 1.2 Hz, 1 H), 5.19 (dd,
J = 17.1, 1.2 Hz, 1 H), 5.84 (ddt,
J = 17.1, 10.5, 5.6 Hz, 1H);
13C NMR
pdf (101 MHz, CDCl
3) δ: 43.6, 52.4, 116.2, 134.7, 157.1. MS (EI)
m/z 115 (M
+, 91), 100 (100). Anal. Calcd. for C
5H
9NO
2: C, 52.16; H, 7.88; N, 12.17. Found: C, 51.77; H, 7.89; N, 11.89.
7.
1,3-Decadiyne was prepared via the
Organic Syntheses procedure directly preceding this procedure. Acetone (99.5%) was purchased from Aldrich Chemical Company and was used as received.
8.
N-Bromosuccinimide (NBS) (99%) was purchased from Aldrich Chemical Company, Inc. and purified by recrystallization from water according to the following procedure.
2 A
500-mL Erlenmeyer flask equipped with a magnetic stir bar was charged with NBS (30 g, 169 mmol) and 300 mL of deionized water and heated on a hot plate until the yellow mixture became homogeneous (approximately 90 °C). The resulting dark orange solution was cooled in an ice bath over 3 h and the white crystals that formed were collected by vacuum filtration on a Büchner funnel, washed with 100 mL of water, and then dried over P
2O
5 in a vacuum dessicator at 22 mmHg for 12 h to afford 21.2 g of NBS as white crystals.
9.
Silver(I) nitrate (99.9%) was obtained from Alfa Aesar and used as received.
10.
The reaction mixture was shielded from light due to the photosensitivity of NBS and bromodiyne
4.
11.
The progress of the reaction was monitored by thin layer chromatography. TLC analysis was conducted using silica gel plates (elution with hexanes, visualization by UV and vanillin); R
f values: diyne
3 = 0.41, bromodiyne
4 = 0.49.
12.
Washing with saturated sodium thiosulfate solution removed traces of bromine.
13.
The product exhibits the following spectroscopic properties: R
f = 0.49 (hexanes); IR (neat) cm
−1: 2956, 2930, 2858, 2141, 1466;
1H NMR
pdf (400 MHz, CDCl
3) δ: 0.89 (t,
J = 7.1 Hz, 3 H), 1.24–1.32 (m, 4 H), 1.38 (app quint,
J = 7.1 Hz, 2 H), 1.52 (app quint,
J = 7.2 Hz, 2 H), 2.24 (t,
J = 7.0 Hz, 2 H);
13C NMR
pdf (125 MHz, CDCl
3) δ: 14.2, 19.2, 22.7, 28.2, 28.7, 31.4, 37.0, 65.5, 65.9, 77.7. Anal. Calcd. for C
10H
13Br: C, 56.36; H, 6.15. Found: C, 56.35; H, 6.29.
14.
In contrast to many other alkynyl bromides we have worked with, the bromodiyne
4 does not exhibit lachrymatory properties. As with all alkynyl halides, however, we nonetheless recommend avoiding any skin contact with this compound and carrying out all operations in a fume hood.
15.
Tetrahydrofuran was purified by pressure filtration through activated alumina under argon.
16.
Pyridine was purchased from Aldrich Chemical Company and purified by distillation under argon from CaH
2. The submitters found that lower yields were obtained in these coupling reactions if pyridine (Alfa Aesar, 99%, 0.1% water content) from a freshly opened bottle was used instead (see Discussion Section).
17.
Potassium hexamethyldisilazide solution was purchased from Alfa Aesar and used as received. KHMDS in toluene (0.5 M, Aldrich) can be employed with similar results.
18.
Copper(I) iodide (98%) was purchased from Alfa Aesar and purified according to the following procedure. A
500-mL, round-bottomed flask was fitted with a Soxhlet extractor equipped with a reflux condenser fitted with a rubber septum and argon inlet needle. The reaction apparatus was flame-dried under vacuum (0.08 mmHg) and purged with argon. A Soxhlet thimble filled with 25 g of CuI was inserted into the extractor and 200 mL of THF was added via syringe. The extractor was wrapped with aluminum foil to shield the CuI from light and the THF was heated in an oil bath at reflux for 24 h. The apparatus was allowed to cool to room temperature and the CuI was transferred to a flame-dried,
100-mL, one-necked, round-bottomed flask and dried at 0.08 mmHg for 24 h. The resulting CuI was ground up with glass rod to afford a white, free-flowing powder, which was stored under argon shielded from light.
19.
The yield of ynamide product in a related coupling reaction was unchanged when CuI was used as received without purification.
20.
Increasing the rate of addition resulted in reduced yields (see Discussion Section).
21.
The progress of the reaction was monitored by thin layer chromatography. TLC analysis was conducted using silica gel plates (elution with 20% EtOAc-hexanes, visualization by UV and vanillin or KMnO
4); R
f values: bromodiyne
4 = 0.69, carbamate
2 = 0.17, diynamide
5 = 0.48.
22.
The aqueous phase in the fourth wash should be colorless. Extraction of copper salts from the organic phase was found to be important in order to obtain the ynamide product in good yield.
23.
The yield of ynamide varied from 73–80% in several runs carried out between one-half-scale and the scale described here.
24.
The product exhibits the following spectroscopic properties: R
f = 0.48 (hexanes/EtOAc, 4/1); IR (neat) cm
−1: 2956, 2261, 2170, 174, 1442;
1H NMR
pdf (400 MHz, CDCl
3) δ: 0.88 (t,
J = 7.0 Hz, 3 H), 1.23–1.32 (m, 4 H), 1.38 (app quint,
J = 7.2 Hz, 2 H), 1.52 (app quint,
J = 7.1 Hz, 2 H), 2.29 (t,
J = 7.1 Hz, 2 H), 3.82 (s, 3 H), 4.05 (d,
J = 6.1 Hz, 2 H), 5.25 (dd,
J = 1.2, 10.3 Hz, 1 H), 5.29 (dd,
J = 1.3, 17.3 Hz, 1 H), 5.85 (ddt,
J = 6.0, 10.3, 18.8 Hz, 1 H);
13C NMR
pdf (101 MHz, CDCl
3) δ: 14.3, 19.8, 22.7, 28.5, 28.7, 31.5, 52.7, 54.5, 58.0, 64.3, 67.5, 84.3, 119.3, 131.3, 155.9; MS (EI)
m/z: 247 (M
+, 100). Anal. Calcd. for C
15H
21NO
2: C, 72.84; H, 8.56; N, 5.66. Found: C, 72.59; H, 8.40; N, 5.31.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
In recent years, ynamides have emerged as exceptionally useful building blocks for organic synthesis.
3 Considerably more robust than simple ynamines, ynamides are more easily stored and handled, and more resistant to hydrolysis and polymerization. Ynamides tolerate a variety of reaction conditions incompatible with simple ynamines and have thus proved to be versatile substrates for a variety of synthetic transformations. Recent representative examples include Pauson-Khand reactions,
4 thermal and metal-promoted [4 + 2] cycloadditions,
5 [2 + 2 + 2] cyclotrimerizations,
6 Heck and other carbometalation reactions,
7 ring-closing metatheses,
8 radical-π cyclizations,
9 Pt-catalyzed cycloisomerizations,
10 ketene [2 + 2] cycloadditions,
11 and Pictet-Spengler-type cyclizations.
12Several strategies are available for the synthesis of ynamides.
3 While some success has been realized using the base-catalyzed isomerization of propargyl amides
13 and the dehydrohalogenation of halo enamides,
14 the most attractive approach to the synthesis of ynamides employs the direct
N-alkynylation of amine derivatives. The first effective means for achieving this transformation was based on the addition of metalated amides to alkynyl(phenyl)iodonium salts.
15 Unfortunately, this method is subject to several serious limitations. While
sulfonamide derivatives participate smoothly in these reactions, lactams, acyclic carbamates, and other carboxamide derivatives do not undergo alkynylation in good yield. Most significantly, this strategy can only be applied to the synthesis of ynamides in which the substituent on the alkynyl carbon is a hydrogen atom, or trialkylsilyl or aryl group.
Recent advances in copper-promoted coupling reactions of amine derivatives have provided the basis for a more general and efficient method that is applicable to the synthesis of a wide variety of ynamides. The preparation of diynamide
5 described here illustrates the method introduced by our laboratory in 2003 for the
N-alkynylation of several classes of amine derivatives.
16 In our initial studies, we attempted to effect the coupling of carbamates with alkynyl halides using the conditions developed by Buchwald
17 for the amidation of aryl halides. Unfortunately, attempted application of Buchwald's catalyst system to the coupling of acyclic carbamates with 1-bromo-2-phenylacetylene gave only trace amounts of the desired ynamide, with the predominant product being the 1,3-diyne generated from "homocoupling" of the alkynyl halide. After considerable experimentation, success in effecting the desired alkynylation was finally achieved when we turned our attention to protocols in which complete conversion of the amide substrate to its copper derivative was carried out
prior to addition of the alkynyl halide.
18 Under these conditions, the rate of alkynylation is significantly faster, copper-promoted dimerization of the alkynyl halide is greatly diminished, and the desired ynamides emerge as the major product of the reaction.
Both alkynyl bromides and alkynyl iodides participate smoothly in the desired reaction.
19 The procedure described here for the preparation of 1-bromo-1,3-decadiyne is based on the method of Hofmeister and coworkers
20 and provides the alkynyl bromide in near-quantitative yield and in a state of high purity. If extended storage is required, we recommend storing this compound as a solution in pentane at −20 °C.
The alkynylation procedure described here is a slightly modified version of our original protocol. As reported previously, a solution of the amide derivative is first deprotonated by reaction with 1 equiv of commercially available KHMDS (solution in THF) and then treated with 1 equiv of CuI. Somewhat diminished yields result if CuCN is employed in place of CuI. Although a full equivalent of CuI is required for the reaction, this salt is inexpensive and can be used as received without purification. After 2 h, the alkynyl bromide is added dropwise and the reaction is allowed to proceed at room temperature overnight. Slow addition of the bromide is important in minimizing the homocoupling side reaction. Only a slight excess of 4 (1.15 equiv) is employed in the optimal procedure described here, although in the case of other alkynyl bromides we occasionally found that the yield of coupling product is improved if 1.5 or even 2 equiv of the alkyne is employed. Originally we carried out the alkynylation reactions in a mixture of pyridine, benzene (from the alkynyl bromide stock solution), and THF (60:30:10 ratio by volume). In the optimized procedure described here, however, the amount of pyridine has been reduced to 25 equivalents, and the final solvent system consists of a mixture of THF and pyridine in a ratio of approximately 3:1.
The examples shown in Table 1 illustrate the scope of this method for the synthesis of ynamides. As reported previously, a broad range of alkynyl bromides participate in the reaction, including compounds that are especially prone to homocoupling such as halo derivatives of conjugated enynes and aryl acetylenes. Acyclic carbamates, sulfonamides, oxazolidinones, and cyclic ureas all undergo N-alkynylation in good yield. Significantly, the alkynylation reaction proceeds smoothly at room temperature, allowing its application to the synthesis of several classes of thermally sensitive ynamides, which are not available by other methods that require reaction at elevated temperature (vide infra).
Concurrent with our studies, Hsung and co-workers developed a complementary method for the synthesis of ynamides that also involves the coupling of amine derivatives with halo acetylenes. In 2003, Hsung reported that the copper catalyst system originally developed by Buchwald for the
arylation and
vinylation of amides, lactams, and carbamates
17 is also effective for the alkynylation of oxazolidinones and lactams.
21 The Hsung procedure requires reaction at elevated temperatures (110–150 °C) and employs catalytic CuCN in conjunction with a 1,2-diamine ligand in the presence of K
3PO
4. Unfortunately, under these conditions acyclic carbamates, sulfonamides, and cyclic ureas undergo alkynylation in poor yield. In 2004, Sato and co-workers reported a modified Buchwald protocol that is applicable to the alkynylation of acyclic and cyclic sulfonamide derivatives,
22 and the same year Hsung described a “second-generation catalyst system”
23 employing catalytic copper sulfate and 1,10-phenanthroline as ligand that is applicable to a broad range of amine derivatives including sulfonamides, imidazolidinones, and acyclic carbamates. This procedure does require reaction at elevated temperature (60–95 °C), however, and therefore is not applicable to the preparation of thermally sensitive ynamides. In the case of ynamide
5, we found that the desired diynamide is produced in only 34–38% yield using the Hsung “second-generation” protocol as compared to 74–82% employing our room temperature procedure.
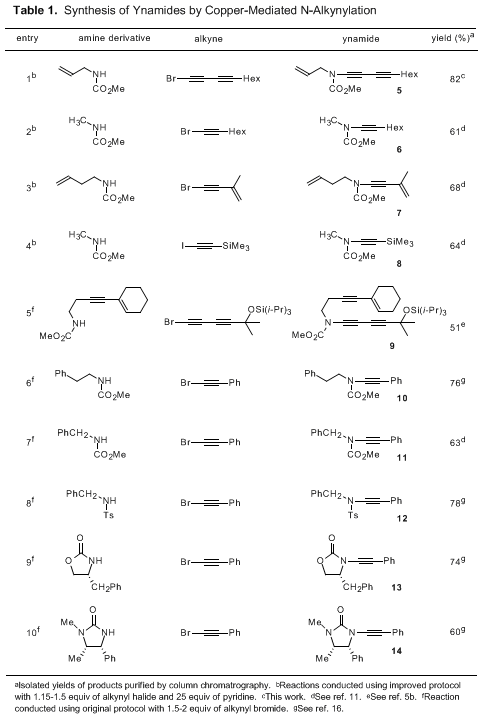
For the synthesis of most ynamides, we have found both our method and the Hsung "second-generation" procedure to be reliable and reproducible on both small and large (i.e., multigram) scales. Recently, Tam and co-workers reported an alternative
N-alkynylation protocol
24 that involves a melding of the procedures previously developed in our laboratory and that of Hsung. Tam's conditions involve the use of catalytic (8–40 mol%) CuI and 1,10-phenanthroline (12–46 mol%), with slow addition of 1.0–1.7 equiv of KHMDS over several hours to a hot (90 °C) solution of the copper salt, ligand, amine derivative, and alkynyl bromide in toluene. Tam was motivated to introduce this modified procedure when he encountered difficulties in reproducing results previously reported by both our group and that of Hsung. For example, Tam reports obtaining none of the desired ynamide from the reaction of 1-bromo-2-phenylacetylene with BnNHCO
2Me using either our method or the method of Hsung. Hsung had previously reported obtaining this ynamide in 73% yield,
23 and we had reported the synthesis of the corresponding Boc derivative in 61% yield.
16We investigated the coupling of BnNHCO
2Me with 1-bromo-2-phenylacetylene in some detail in an attempt to identify the experimental variables that might be responsible for Tam's inability to reproduce our results and the results of Hsung and co-workers.
11 We obtained the expected ynamide
11 in 63% yield using our method (Table 1, entry 7), and in 79% yield by employing the “second-generation” procedure of Hsung. In our experience, both methods have proved to be highly reproducible. We have noted, however, that results employing our method are affected by the quality of the pyridine used in the reaction. Thus, the yield reported here for ynamide
11 was obtained using pyridine freshly distilled from CaH
2 or KOH as recommended in our original report.
16 When pyridine (Alfa Aesar, 99%, 0.1% water content) from a freshly opened bottle was used (without distillation) instead, the yield of ynamide product declined to 52%. Most significantly, when distilled pyridine that had then been exposed to the atmosphere for several days was used for the
N-alkynylation, none of the desired ynamide was formed.
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Allylamine:
2-Propen-1-amine; (107-11-9)
Methyl chloroformate:
Carbonochloridic acid,
methyl ester; (79-22-1)
N-(Methoxycarbonyl)-2-propenylamine:
Carbamic acid, 2-propenyl-,
methyl ester; (19364-21-7)
1,3-Decadiyne; (55682-66-1)
N-Bromosuccinimide:
2,5-Pyrrolidinedione,
1-bromo-; (128-08-5)
Potassium hexamethyldisilazide:
Silanamine,
1,1,1-trimethyl-N-(trimethylsilyl)-,
potassium salt; (40949-94-8)
Copper(I) iodide; (7681-65-4)
 |
Rick L. Danheiser received his undergraduate education at Columbia where he carried out research in the laboratory of Professor Gilbert Stork. He received his Ph.D. at Harvard in 1978 working under the direction of E. J. Corey on the total synthesis of gibberellic acid. Dr. Danheiser is the A. C. Cope Professor of Chemistry at MIT where his research focuses on the design and invention of new annulation and cycloaddition reactions, and their application in the total synthesis of biologically active compounds. |
 |
Amanda L. Kohnen was born in 1981 in West Hollywood and grew up in Yuma, Arizona. She did her undergraduate work at the University of California, San Diego and received a B.S. degree in Chemistry in 2004. She is now pursuing graduate studies at the Massachusetts Institute of Technology where her research under the direction of Professor Rick Danheiser involves the development of new synthetic methods and the total synthesis of biologically active natural products. Ms. Kohnen is the recipient of an MIT Leventhal Presidential Fellowship, a Pfizer Research Fellowship, and an NSF Graduate Research Fellowship. |
 |
Joshua R. Dunetz received his Ph.D. from MIT in 2005 after completing graduate studies under the direction of Professor Rick L. Danheiser. His graduate research focused on the synthesis of ynamides and their cycloadditions with conjugated enynes in a strategy for the synthesis of indolines and indoles. In other graduate work, Dr. Dunetz studied the synthesis of nitrogen heterocycles in environmentally friendly reaction media such as supercritical carbon dioxide. He is currently a postdoctoral research associate with Professor William Roush at Scripps Florida working on the total synthesis of macrolide natural products. |
 |
Xiaorong Liu graduated with BS and MS degrees in Chemistry from Nanjing University in China where she carried out research on the synthesis of organic conducting polymers in the laboratory of Professor Xue Gi. In the fall of 1999, she began graduate studies under the direction of Professor Robert K. Boeckman, Jr. at the University of Rochester where her PhD thesis focused on the total synthesis of (–)-Apoptolidin. In the fall of 2004, she joined the laboratory of Professor Denmark as a postdoctoral associate at University of Illinois at Urbana-Champaign. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved