Org. Synth. 2009, 86, 47
DOI: 10.15227/orgsyn.086.0047
(S)-(-)-2-ALLYLCYCLOHEXANONE
[2-Allylcyclohexan-1-one]
Submitted by Manfred Braun, Panos Meletis, and Mesut Fidan
1.
Checked by Ye Zhu and Viresh H. Rawal.
1. Procedure
A 1-L round-bottomed flask (Note 1) equipped with a 3-cm egg-shaped magnetic stir bar is charged with tris(dibenzylideneacetone)-dipalladium(0)-chloroform adduct (0.388 g, 0.375 mmol, 0.005 equiv), (S)-(-)-5,5'-dichloro-6,6'-dimethoxy-2,2'-bis(diphenylphosphino)-1,1'-biphenyl (0.977 g, 1.50 mmol, 0.02 equiv) and lithium chloride (7.63 g, 180 mmol, 2.4 equiv) (Note 2). The flask is closed with a rubber septum, connected to a combined nitrogen/vacuum line via a 16-gauge needle, evacuated (22 °C, 0.4 mmHg) for 5 h in order to remove traces of water from the lithium salt, and filled with nitrogen. To this flask is added a solution of allyl methyl carbonate (8.52 mL, 8.71 g, 75.0 mmol, 1.00 equiv) (Note 3) in dry tetrahydrofuran (THF) (195 mL) (Note 4) over 5 min through a 16-gauge cannula (Note 5). The deep purple homogeneous solution is stirred at 22 °C for 1 h. In the course of stirring, the color changes to yellow. The resulting mixture is cooled down to -78 °C in a dry ice/acetone bath with stirring. A 500-mL, three-necked, round-bottomed flask is equipped with a 3-cm egg-shaped magnetic stir bar, a glass stopper, a rubber septum fitted with the combined nitrogen/vacuum line via a 16-gauge needle, and a low-temperature thermometer via a thermometer adapter (Note 6). The flask is evacuated and refilled with nitrogen three times. To this flask, diisopropylamine (11.1 mL, 7.97 g, 78.7 mmol, 1.05 equiv) (Note 7) is injected by syringe, and dry THF (75 mL) is added via a 16-gauge cannula. The flask is immersed in a dry ice/acetone bath and allowed to stir for 30 min. A 1.60 M solution of n-butyllithium in n-hexane (46.9 mL, 4.80 g, 75.0 mmol, 1.00 equiv) (Note 8) is added dropwise by syringe over 30 min. In the course of the addition, the internal temperature of the solution should not exceed -70 °C. The dry ice/acetone bath is replaced by an ice bath, and stirring is continued for 30 min. The mixture is then recooled in the dry ice/acetone bath, and a solution of freshly distilled cyclohexanone (7.78 mL, 7.36 g, 75.0 mmol, 1.00 equiv) (Note 9) in dry THF (75 mL) is added dropwise by a syringe over 1 h. In the course of the addition, the internal temperature of the solution should not exceed -70 °C. The mixture is stirred in an ice bath for 30 min and cooled again to -78 °C (internal temperature) in a dry ice/acetone bath. This colorless solution is then transferred over 10 min through a 16-gauge cannula into the 1 L flask (Note 5). After stirring for 40 h at -78 °C (Note 10), a yellow solution with white precipitate is obtained. This mixture is poured rapidly into a 4 L Erlenmeyer flask charged with 1.5 L of phosphate-buffered water (pH 7.00) (Note 11) stirred by a 7-cm egg-shaped magnetic stir bar. The resulting mixture is transferred into a 4 L separatory funnel, and extracted with three portions (170 mL, 170 mL, and 160 mL) of dichloromethane (CH2Cl2) (Note 11). The combined organic layers are dried over 20 g magnesium sulfate (Note 12), filtered, and concentrated by rotary evaporation (30 °C, 15 mmHg). The resulting brown-colored crude product is purified by Kugelrohr distillation (Note 13). A 50-mL pear-shaped flask containing the crude product is heated to 50 °C, and a 100-mL receiving flask is cooled in an ice-water bath. While maintaining a pressure of 0.05 mmHg, the pure product is collected as a colorless liquid; 8.08 g (78 %). The enantiomer ratio is determined by GC; ee: 94 % (Notes 14, 15).
2. Notes
1.
The apparatus was oven-dried overnight, assembled hot and maintained under a positive pressure of nitrogen during the course of the reaction.
2.
Tris(dibenzylideneacetone)dipalladium(0) chloroform adduct was obtained from Strem Chemicals, Inc. The submitters noted that distinctly lower enantioselectivity was obtained when the reagent of other suppliers was used. (
S)-(-)-5,5'-Dichloro-6,6'-dimethoxy-2,2'-bis(diphenylphos-phino)-1,1'-biphenyl (min. 95 %) was also purchased by the checkers from Strem Chemicals, Inc. The submitters obtained the ligand from LANXESS. Lithium chloride (SigmaUltra, min. 99.0 %) was obtained from Sigma-Aldrich, Inc. The submitters obtained lithium chloride (Normalpur) from VWR-Prolabo.
3.
Allyl methyl carbonate is commercially available from Sigma-Aldrich, Inc. The submitters prepared it according to: H. O. L. Fischer, L. Feldmann,
Ber. Dtsch. Chem. Ges. 1929,
62, 854. The checkers prepared it according to: I. Minami, J. Tsuji,
Tetrahedron 1987,
43, 3903.
4.
Tetrahydrofuran (Oprima
®), was purchased from Fisher Scientific, Inc. It was purged with nitrogen and dried over activated alumina. The submitters purchased Tetrahydrofuran (technical grade) from Kraemer und Martin GmbH. It was refluxed over potassium hydroxide and distilled. Then the distillate was refluxed over sodium wire and distilled under nitrogen. Freshly distilled tetrahydrofuran was taken from the receiving flask via syringe or cannula.
5.
The cannula was placed with one end immersed in the solution to be transferred and the other end in the receiver. A positive nitrogen pressure was maintained over the solution to be transferred, and the receiver was evacuated such that the solution flowed into the receiver at a steady rate. For a description of the technique, see: A. Salzer, in
Synthetic Methods of Organometallic and Inorganic Chemistry, W. A. Herrmann, G. Brauer, Eds.; Vol. 1, W. A. Herrmann, A. Salzer, Volume Eds.; Thieme, Stuttgart
1996, page 26.
6.
The submitters used a thermocouple connected to a resistance thermometer introduced through a septum to indicate the internal temperature.
7.
Diisopropylamine (purity >99.5 %) was purchased from Sigma-Aldrich, Inc. It was refluxed over calcium hydride and distilled under nitrogen. The submitters used 1.00 equiv
(7.59 g, 10.5 mL, 75.0 mmol) of diisopropylamine.
8.
n-Butyllithium (1.6 M solution in n-hexane) was purchased from Sigma-Aldrich, Inc., and titrated according to:
J. Org. Chem. 1976,
41, 1879. The submitters purchased
n-butyllithium (1.6 M solution in
n-hexane) from Acros Organics.
9.
Cyclohexanone (Selectphore®) was obtained from Sigma-Aldrich, Inc. The submitters obtained cyclohexanone (extra pure) from Riedel-de Haën. It was refluxed over calcium hydride and distilled under nitrogen.
10.
In order to keep the bath temperature at -78 °C, the Dewar vessel was covered with a 3.5 cm thick insulating lid of Styropor
®, cut in such a way that space is left only for the neck of the reaction flask. Solid dry ice was always maintained in the acetone bath. Additional dry ice was to be added every 6-8 hours. The submitters noted that shorter reaction time (15 h) led to a decrease in yield.
11.
Buffer solution pH 7.00 with fungicide was supplied by Riedel-de Haën. Dichloromethane (Certified ACS) was purchased from Ficher Scientific, Inc. The submitters used dichloromethane (technical grade) from Ineos Chlor Ltd.
12.
Magnesium sulfate (Anhydrous Certified) was purchased from Ficher Scientific, Inc. The submitters purchased magnesium sulfate (99% DAC) from Grüssing GmbH.
13.
The submitters purified the product by trap-to-trap distillation. The flask containing the crude product was connected by glass tubes to two traps. The two traps were cooled to 0 °C and -196 °C, respectively, and the flask was heated to 50 °C. While maintaining a pressure of 0.05 mmHg, pure product was collected in the 0 °C-trap as a colorless liquid; 8.18 g (79 %);
ee: 94 %. The trap-to-trap distillation is preferred to a conventional distillation in order to prevent partial racemization of the product; in addition, it provides a more efficient separation from starting materials. The checkers also tested the trap-to-trap distillation on a half-scale reaction. The pure product was isolated as a colorless liquid; 4.05 g (78 %);
ee: 94 %.
14.
GC column: Chiraldex™-beta-DP, 20 m × 0.25 mm × 0.25 μm; column-temperature 80 °C; [(
S)-enantiomer t
R = 23.9 min, (
R)-enantiomer t
R = 25.4 min]. The submitters used different GC conditions; column: FS-Hydrodex-beta-TBDAc, 25m × 0.25 mm; column temperature 90 °C; [(
S)-enantiomer t
R = 17.4 min, (
R)-enantiomer t
R = 20.0 min].
15.
Physical and spectroscopic data are as follows: [α]
D25 -17.4 (chloroform,
c 1); the submitters reported: [α]
D20 -17.0 (chloroform,
c 1); lit.
2g: [α]
D20 -15.8 (methanol,
c 3.0); IR (film): 3076, 2935, 2861, 1711, 1641, 1448, 1432, 1313, 1126, 996, 912 cm
-1;
1H-NMR
pdf (500 MHz, CDCl
3) δ: 1.30-1.42 (m, 1 H), 1.61-1.71 (m, 2 H), 1.81-1.92 (m, 1 H), 1.94-2.09 (m, 2 H), 2.09-2.17 (m, 1 H), 2.25-2.44 (m, 3 H), 2.50-2.58 (m, 1 H), 4.96-5.05 (m, 2 H), 5.76 (dddd,
J = 6.5, 8.0, 10.2, 17.0 Hz, 1 H);
13C NMR
pdf (125 MHz, CDCl
3) δ: 24.9, 27.9, 33.4, 33.7, 42.0, 50.2, 116.2, 136.5, 212.5. Anal. Calcd. for C
9H
14O: C, 78.21; H, 10.21; O 11.58. Found: C, 78.01; H, 10.19; O 11.89. HRMS: Calcd. for C
9H
14ONa
+ (M+Na): 161.093686. Found: 161.093303.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The enantiomeric (
R)- and/or (
S)-2-allylcyclohexanone has been chosen frequently in order to demonstrate the scope of different approaches in stereoselective synthesis. Among them, the allylic alkylation of azaenolates and enamines with chiral auxiliary groups has been used frequently.
2 In addition, access to the enantiomeric 2-allylcyclohexanones has been opened by deracemization through enzymic resolution
3 or formation of inclusion compounds.
4 Prochiral allyl-substituted enol acetates and enol carbonates have been converted into nonracemic 2-allylcyclohexanone by enantioface differentiating enzymatic hydrolysis.
5 Allyl-substituted enolates also lead to 2-allylcyclohexanone in an enantioselective way when treated with stoichiometric amounts of proton sources.
6 The allylation of the lithium enolate of cyclohexanone has been accomplished in the presence of chiral amines used in stoichiometric
7 or substoichiometric amounts.
8The palladium-catalyzed asymmetric allylic alkylation has developed into an exceptionally useful and versatile method for enantioselective carbon-carbon and carbon-heteroatom bond formation.
9 However, carbon nucleophiles have been limited to "soft", stabilized carbanions almost exclusively. Extending the asymmetric allylic substitution to include preformed, nonstabilized metal enolates
10 would significantly enhance its versatility, in particular because it would permit enantioselective alkylation in the homoallylic position. After early approaches of combining metal enolates with allylpalladium complexes,
11 the first enantioselective allylic alkylation of a nonstabilized ketone enolate was presented by Trost and Schroeder in 1999 using the tin enolate of 2-methyl-1-tetralone.
12a Shortly thereafter, we reported the enantioselective and diastereoselective allylation of cyclohexanone through the lithium or magnesium enolates.
13 Additional examples using lithium enolates were also reported.
12c-e,16 More recently, it has been shown that enantioselective palladium-catalyzed allylic alkylations are feasible not only with enol stannanes
12a,b but also with enol silanes,
14 and enamines.
15 An alternative solution to the problem of the enantioselective allylic alkylation of nonstabilized enolates relies on their
in situ generation, using allyl β-ketoesters or allyl enol carbonates.
17 Upon treatment with palladium(0) catalysts, carbon dioxide is liberated, and the enolate anion and the cationic palladium complex thus formed combine to give the allylated ketone. The method has been applied mostly for forming quaternary carbon centers, but was also used for the preparation of 2-allylcyclohexanone, starting from enol carbonate
1 as shown below.
17c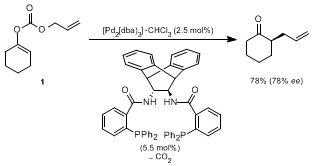
The allylic alkylation through the
in situ generated enolates requires the previous preparation, isolation, and purification of either allyl enol carbonates or β-ketoesters. Compared to our direct allylic alkylation of preformed lithium or magnesium enolates, those protocols can be regarded as a detour in that an additional step is required. Additionally, in the case of 2-allylcyclohexanone, the route through the lithium enolate provides higher enantioselectivity than the enol carbonate route. The direct allylation of lithium and magnesium enolates has been extended to other ketones as well as to substituted allylic starting materials. As shown in Table 1,
18 both enantioselective and diastereoselective variants are possible. The protocol permits the generation of compounds with contiguous stereogenic centers in the allylic and the homoallylic position in an enantioselective and diastereoselective manner (entries 1-3) and can be applied to the asymmetric allylic alkylation of cyclopentanone and 1-tetralone (entries 4 and 5). When extended to diastereomerically and enantiomerically pure allylic substrates (entries 6-9), the achiral ligand bis(diphenylphosphino)ferrocene is suitable to bring about regioselective and diastereoselective allylic alkylations leading to enantiomerically pure products.
Table 1. Enantioselective and/or diastereoselective allylic alkylations of magnesium and lithium enolates.18
Table 1. (continued) Enantioselective and/or diastereoselective allylic alkylations of magnesium and lithium enolates.18
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Tris(dibenzylideneacetone)-dipalladium(0)-chloroform adduct; (52522-40-4)
(S)-(-)-5,5'-Dichloro-6,6'-dimethoxy-2,2'-bis(diphenylphosphino)-1,1'-biphenyl: Phosphine, [(1S)-5,5'-dichloro-6,6'-dimethoxy[1,1'-biphenyl]-2,2'-diyl]bis[diphenyl-; (185913-98-8)
Lithium chloride; (7447-41-8)
Allyl methyl carbonate: Carbonic acid, methyl 2-propen-1-yl ester; (35466-83-2)
Diisopropylamine: 2-Propanamine, N-(1-methylethyl)-; (108-18-4)
n-Butyllithium; (109-72-8)
Cyclohexanone; (108-94-1)
(S)-(-)-2-Allylcyclohexanone: Cyclohexanone, 2-(2-propen-1-yl)-, (2S)-; (36302-35-9)
 |
Manfred Braun, born in 1948 in Schwalbach near Saarlouis, studied chemistry at the University of Karlsruhe from 1966 until 1971, and completed his doctorate under Professor Dieter Seebach in Gießen in 1975. After a postdoc with Professor George H. Büchi at the Massachusetts Institute of Technology in 1975 and 1976, he joined Professor Hans Musso's research group at the University of Karlsruhe and completed his Habilitation there in 1981. Since 1985, he has been a professor of organic chemistry at the Heinrich-Heine-University Düsseldorf. His current research interests include the development of new synthetic methods (especially for asymmetric synthesis), organometallic chemistry, development of metal complexes as smart materials, and syntheses of biologically active compounds. |
 |
Panos Meletis was born 1979 in Neuss, Germany. He graduated in 2006 and received his diploma from the Heinrich-Heine-Universität in Düsseldorf. He prepared his diploma thesis with the topic "Palladium Catalyzed Allylation of Lactone- and Ester-Enolates" under the supervision of Professor M. Braun. In the same year he started his Ph. D. study. His current interest is still the palladium catalyzed allylic alkylation. |
 |
Mesut Fidan was born 1979 in Solingen, Germany. He graduated in 2006 and received his diploma at Heinrich- Heine-Universität in Düsseldorf under the supervision of Professor M. Braun. The topic of his diploma thesis was "Palladium catalyzed allylation with dianions". In the same year he started his Ph.D. study in Organic Chemistry. His current interest is palladium-catalyzed regio- and enantioselective allylation of lactams. |
 |
Ye Zhu was born in Jinan, China in 1983. He received his B.S. in chemistry from Peking University in 2006, and went to the University of Chicago for graduate school in the same year. He is currently a chemistry graduate student with Prof. Viresh Rawal, working on metal-catalyzed and organocatalyzed asymmetric reactions. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved