Org. Synth. 2010, 87, 317
DOI: 10.15227/orgsyn.087.0317
NICKEL-CATALYZED ASYMMETRIC NEGISHI CROSS-COUPLINGS OF RACEMIC SECONDARY ALLYLIC CHLORIDES WITH ALKYLZINCS: (S,E)-ETHYL 6-(1,3-DIOXOLAN-2-YL)-4-METHYLHEX-2-ENOATE
[2-Hexenoic acid, 6-(1,3-dioxolan-2-yl)-4-methyl-, ethyl ester, (2E,4S)-]
Submitted by Sha Lou and Gregory C. Fu
1.
Checked by David I. Chai, Gavin Chit Tsui, and Mark Lautens.
1. Procedure
A. Ethyl (E)-4-hydroxy-2-pentenoate (1). An oven-dried, 1000-mL, two-necked, round-bottomed flask equipped with a rubber septum, an argon inlet, and a magnetic stir bar [41.3 × 19.0 mm, egg-shaped] is purged with argon and charged with Et2O
(500 mL) (Note 1) and ethyl (E)-4-oxo-2-butenoate (24.1 mL, 25.6 g, 200 mmol, 1.0 equiv) (Note 2) by syringe. The solution is cooled to -78 °C in a dry ice/acetone bath, and then MeMgBr (3.0 M in Et2O; 70.0 mL, 210 mmol, 1.05 equiv) (Note 3) is added dropwise via syringe pump (2.0 mL/min) over 35 min (the slow addition can also be conducted via a 100-mL pressure-equalizing addition funnel fitted with a septum). After the addition is complete, the reaction mixture is stirred for an additional 1 h at -78 °C. Additional MeMgBr (3.0 M in Et2O; 60.0 mL, 180 mmol, 0.90 equiv) (Note 3) is added dropwise via syringe pump (2.0 mL/min) over 35 min, and the reaction mixture is stirred for an additional 1 h at -78 °C. During the course of the reaction, the precipitation of white solids leads to a heterogeneous reaction mixture. The progress of the reaction can be followed by TLC analysis on SiO2 (60% Et2O/pentane as the eluent; visualization with a UV lamp or with a KMnO4 stain; the aldehyde starting material has an Rf = 0.8 and the alcohol product has an Rf = 0.4) (Note 4). Then, the dry ice/acetone bath is removed, and the reaction mixture is carefully poured over 5 min into a rapidly stirred, 0 °C saturated aqueous solution of NH4Cl
(300 mL) in a 2-L Erlenmeyer flask equipped with a magnetic stir bar. The two-necked flask is washed with a saturated aqueous solution of NH4Cl
(100 mL) and then Et2O
(100 mL). The aqueous solution and the organic solution are combined and transferred to a 2-L separatory funnel. The aqueous phase is separated and extracted with Et2O (200 mL × 3). The organic layers are combined, dried over MgSO4 (30 g), and filtered through a Büchner funnel containing a bed of celite (1.0 cm height). The filtrate is concentrated by rotary evaporation (30 mmHg). The residue is loaded onto a wet-packed (hexanes) column of SiO2 (8 cm diameter × 22 cm height; 250 g) and eluted with a gradient of Et2O in hexanes (500 mL of hexanes; 1.0 L of 15% Et2O/hexanes, 1.0 L of 20% Et2O in hexanes, 1.0 L of 30% Et2O/hexanes, 1.0-2.0 L of 50% Et2O/hexanes; fraction size: 100 mL) (Note 5) (TLC analysis on SiO2: 60% Et2O/pentane as eluent, visualization with a UV lamp and a KMnO4 stain, Rf = 0.4) (Note 4). The desired allylic alcohol 1(22.3 g, 77% yield) is obtained as a yellow oil (Note 6).
B. Ethyl (E)-4-chloropent-2-enoate (2). An oven-dried, 500-mL, two-necked, round-bottomed flask equipped with a rubber septum, an argon inlet, and a magnetic stir bar [31.8 × 15.9 mm, egg-shaped] is purged with argon and charged with CH2Cl2 using a 50-mL syringe (200) (Note 7) and ethyl (E)-4-hydroxy-2-pentenoate (1) (14.4 g, 100 mmol, 1.00 equiv). The solution is cooled to 0 °C in an ice bath, and dichlorotriphenylphosphorane (40.0 g, 120 mmol, 1.20 equiv) (Note 8) is added, followed by imidazole (8.17 g, 120 mmol, 1.20 equiv) (Note 9). The solution is allowed to warm to rt, and then it is stirred for 6 h, during which time a white precipitate forms. The progress of the reaction can be followed by TLC analysis on SiO2(50% Et2O/pentane as the eluent; visualization with a UV lamp or with a KMnO4 stain; the alcohol starting material has an Rf = 0.2, and the chloride product has an Rf = 0.8) (Note 4). Pentane (150) and ether (150) are poured into the reaction flask slowly over 3 min, and the white precipitate is removed by filtration through SiO2(8 cm diameter × 10 cm height). The SiO2is washed with pentane/ether (1/1; 400 mL). The combined organic layers are concentrated by rotary evaporation (30 mmHg), and the residue is transferred to a 50-mL round-bottomed flask equipped with a magnetic stir bar. The product is distilled under reduced pressure (16-20 mmHg, 75-80 °C) through a short-path distillation head to afford the desired allylic chloride (2) (13.2 g, 82% yield) as a colorless oil (Note 10).
C. 2-[2-(1,3-Dioxolan-2-yl)ethyl]zinc bromide (3). An oven-dried, 200-mL, two-necked, round-bottomed flask equipped with a rubber septum and a magnetic stir bar [15.9 × 9.5 mm, octagon-type] is purged with argon for 5 min and charged with zinc powder (9.80 g, 150 mmol, 1.50 equiv) (Note 11). The zinc is heated under vacuum (0.5 mmHg) at 70 °C in an oil bath for 30 min. Anhydrous N,N-dimethylacetamide (DMA; 100 mL) (Note 12) is added using a 50-mL syringe, and I2 (634 mg, 2.50 mmol, 0.0250 equiv) (Note 13) is added in one portion via the side neck of the round-bottomed flask. The red-gray solution is stirred at 70 °C until the red color fades (~5 min). 2-(1,3-Dioxolan-2-yl)ethyl bromide (12.0 mL, 100 mmol, 1.00 equiv) (Note 14) is added via syringe over 3 min, and then the reaction mixture is stirred at 70 °C for 12 h. Next, the mixture is allowed to cool to rt, and the unreacted zinc powder is removed by filtration under argon (see Figure 1). The clear yellow solution is employed in the next step without further purification. 1H NMR spectroscopy with a D2O insert is used to determine that the concentration of the alkylzinc solution is 0.697 M (Note 15).
Figure 1.
D. (S,E)-Ethyl 6-(1,3-dioxolan-2-yl)-4-methylhex-2-enoate(4). An oven-dried, 1000-mL, three-necked, round-bottomed flask equipped with a thermometer inlet and thermometer, an argon inlet, a rubber septum, and a magnetic stir bar [41.3 × 19.0 mm, egg-shaped] is purged with argon for 20 min. Under a positive pressure of argon, anhydrous NaCl (10.4 g, 177 mmol, 4.00 equiv) (finely ground and dried at 120 °C under vacuum overnight) (Note 16 and Note 17) is added, followed by NiCl2glyme (485 mg, 2.22 mmol, 0.0500 equiv) (Note 18) and (R)-CH2CH2Ph-Pybox (1.04 g, 2.44 mmol, 0.0550 equiv) (Note 19 and Note 20). Anhydrous DMF (222) (Note 21) and anhydrous DMA (155) (Note 22) are then added via 50-mL syringe, and the flask is capped with a rubber septum. This solution of the catalyst is stirred at 0 °C for 20 min in an isopropanol bath cooled by a chiller system (Note 23). Ethyl (E)-4-chloropent-2-enoate (2) (7.20 g, 44.3 mmol, 1.00 equiv) is added via 50-mL syringe, and then a solution of 2-[2-(1,3-dioxolan-2-yl)ethyl]zinc bromide 3 (75.7 mL, 0.70 M in DMA, 53 mmol, 1.2 equiv) is added via another 50-mL syringe over 10 min. Throughout the addition of the alkylzinc reagent, the temperature of the solution is maintained at 0 °C. The reaction mixture is stirred for 24 h at 0 °C. The progress of the reaction can be followed by GC analysis (Note 24). After the reaction is complete, water
(400 mL) is added over 20 min to quench the excess organozinc reagent, during which time the temperature of the reaction mixture is kept below 25 °C. After the addition of water is complete, the solution is stirred at rt for an additional 10 min, and then it is extracted with Et2O (200 mL × 3). The combined organic layers are washed with water (200 mL × 2), dried over anhydrous Na2SO4 (30 g), filtered through a Büchner funnel containing a bed of celite (1.0 cm height), and concentrated by rotary evaporation (30 mmHg). The residue is loaded onto a wet-packed (hexanes) column of SiO2 (4 cm diameter × 30 cm height; 350 g) and eluted with a gradient of Et2O in hexanes (500 mL of hexanes, 2.0 L of 5% Et2O/hexanes, 2.0 L of 10% Et2O/hexanes, 2.0 - 4.0 L of 15% Et2O/hexanes) (Note 5) (TLC analysis on SiO2: 50% Et2O/pentane as eluent, visualization with a KMnO4 stain, Rf = 0.5) (Note 4). The desired cross-coupling product 4 (9.0 g, 89% yield, 97% ee) is obtained as a colorless oil (Note 24).
2. Notes
1.
Et2O (99+%) was purchased from Caledon and purified by passage through activated alumina under nitrogen.
2.
Ethyl (E)-4-oxo-2-butenoate (96%) was purchased from Alfa Aesar and used as received.
3.
MeMgBr (3.0 M in Et
2O) was purchased from Aldrich and used as received.
4.
Analytical thin-layer chromatography was performed using EMD 0.25 mm silica gel 60-F plates.
5.
Column chromatography was performed on Silicycle 60 Å silica gel.
6.
On half-scale, compound
1was isolated in 80% yield (11.6 g); Compound
1 has the following physical properties:
1H NMR
pdf (CDCl
3, 400 MHz) δ: 1.30 (t,
J = 7.2 Hz, 3 H), 1.34 (d,
J = 6.8 Hz, 3 H), 2.03 (br s, 1 H), 4.20 (q,
J = 6.8 Hz, 2 H), 4.46-4.49 (m, 1 H), 6.00 (dd,
J = 15.6, 1.6 Hz, 1 H), 6.95 (dd,
J = 15.6, 4.8 Hz, 1 H);
13C NMR
pdf (CDCl
3, 100 MHz) δ: 14.2, 22.7, 60.5, 67.2, 119.6, 151.0, 166.7. The spectral data are in agreement with the reported values.
2 The purity (95%) was determined by GC analysis (t
r = 2.61 min; Agilent Technologies 7890A Series GC system equipped with a DB-1 column (length 30 m, I.D. 0.25 mm, film 0.25 μm); carrier gas: H
2 (2 mL/min); gradient temperature: hold at 100 °C for 1 min, 100 → 280 °C at 40 °C/min, hold at 280 °C for 2 min).
7.
Dichloromethane (>99.8%) was purchased from ACP Chemicals and purified by passage through activated alumina under nitrogen.
8.
Dichlorotriphenylphosphorane (95%) was purchased from Aldrich and used as received.
9.
Imidazole (99%) was purchased from Alfa Aesar and used as received.
10.
On half-scale, compound
2 was isolated in 85% yield (13.7 g); compound
2 has the following physical properties: IR (neat) 2982, 2932, 2905, 1724, 1658, 1446, 1369, 1342, 1273, 1226, 1180, 1033, 1015, 976 cm
-1;
1H NMR
pdf (CDCl
3, 400 MHz) δ: 1.30 (t,
J = 7.2 Hz, 3 H), 1.64 (d,
J = 6.4 Hz, 3 H), 4.22 (q,
J = 6.8 Hz, 2 H), 4.63 (d of pentets,
J = 6.8, 1.2 Hz, 1 H), 6.00 (dd,
J = 15.6, 1.2 Hz, 1 H), 6.93 (dd,
J = 15.2, 6.8 Hz, 1 H);
13C NMR
pdf (CDCl
3, 100 MHz) δ: 14.2, 24.3, 54.8, 60.8, 121.8, 147.0, 165.9; GCMS
m/z calcd. for C
7H
11ClO
2 (M
+) 162.0448; found 162.0447. The purity (96%) was determined by GC analysis (t
r = 2.51 min), using the same conditions as for compound
1.
11.
Zinc powder (99.9%, ~100 mesh) was purchased from Alfa Aesar and used as received.
12.
Anhydrous DMA (99.8%) purchased from Aldrich (water content: 14 ppm) wasused as received.
13.
I2 (≥99%, chips) was purchased from Aldrich and used as received.
14.
2-(1,3-Dioxolan-2-yl)ethyl bromide (95%) was purchased from Alfa Aesar and used as received.
15.
For the NMR experiment, a
1H NMR spectrum of the organozinc solution with a D
2O insert (prepared in a melting point capillary, and sealed with flame) was taken to determine the concentration of the solution. The resonances for DMA appear at δ 3.02, 2.84, and 2.02. The resonance for the CH
2Zn protons of RZnBr appears at δ 0.00. The resonance for the terminal Me group of the reduction product appears at δ 0.87. The molar ratio of DMA : RZnBr : RH (X : 1 : Y) was determined by No-D NMR to be 15.12 : 1 : 0.20. The density (d) of the solution was measured to be 1.10 g/mL. The final concentration of alkylzinc was calculated to be 0.697 M according to the equation: [RZnBr] = d × 1000/(X × MW
DMA + MW
RZnBr + Y × MW
RH); For half-scale, X = 16.457, Y = 0.317, d = 1.17 g/mL, and the concentration of alkylzinc was 0.68 M.
16.
NaCl (99.999%) was purchased from Aldrich, ground to a fine powder and dried under vacuum at 150 °C overnight before use. The use of anhydrous NaCl beads (99.99% from Aldrich) or NaCl (99.5% from Mallinckrodt Chemicals) powder that was oven-dried in the air furnished lower yields (30% and 74%, respectively).
17.
The presence of NaCl has a substantial impact on the rate of the cross-coupling, but little impact on the ee. The role of the NaCl may be to increase the ionic strength of the reaction mixture (the use of more polar solvents is generally advantageous) or to activate the organozinc reagent.
318.
NiCl
2•glyme was purchased from Strem and used as received.
19.
(
R)- or (
S)-CH
2CH
2Ph-Pybox can be prepared via the reaction of 2-amino-4-phenylbutan-1-ol with 2,6-pyridinedicarbonitrile catalyzed by 5% zinc triflate in toluene at reflux.
420.
(
S)-CH
2CH
2Ph-Pybox (97%) may be purchased from Aldrich and used as received.
21.
Anhydrous
DMF (≥99.5%, over molecular sieves) was purchased from Aldrich (water content: <0.005%) and used as received.
22.
When anhydrous DMA (99.8%) purchased from Aldrich (water content: 14 ppm) was used as received, the cross-coupling product was obtained in good yield (>90%). However, the use of DMA with a higher water content (e.g, 77 ppm) led to a lower yield of product (~75%).
23.
When the reaction was conducted at -10 °C on a 10-g scale, the cross-coupling proceeded more slowly, resulting in a 52% yield after 24 h.
24.
For half-scale, compound
4was isolated in 90% yield (4.54 g); The product was generated with >20:1 E:Z selectivity and >20:1 regioselectivity. Compound
4 has the following physical properties: [α]
26D +23.6 (
c 1.67, CHCl
3); IR (neat) 2958, 2931, 2878, 1716, 1651, 1458, 1408, 1369, 1265, 1203, 1180, 1142, 1037, 988, 945, 887, 864 cm
-1;
1H NMR
pdf (CDCl
3, 400 MHz) δ: 1.07 (d,
J = 6.8 Hz, 3 H), 1.29 (d,
J = 7.2 Hz, 3 H), 1.48-1.54 (m, 2 H), 1.61-1.68 (m, 2 H), 2.34 (septet,
J = 7.2 Hz, 1 H), 3.81-3.89 (m, 2 H), 3.92-4.00 (m, 2 H), 4.18 (q,
J = 7.2 Hz, 2 H), 4.84 (br t,
J = 4.4 Hz, 1 H), 5.75 (d,
J = 16.0 Hz, 1 H), 6.84 (dd,
J = 16.0, 8.0 Hz, 1 H);
13C NMR
pdf (CDCl
3, 100 MHz) δ: 14.3, 19.4, 30.0, 31.5, 36.4, 60.2, 64.9, 104.4, 120.2, 153.8, 166.8; GCMS
m/z calcd. for C
12H
19O
4 (M
+) 227.1283; found 227.1286. The ee was determined by HPLC analysis with an Agilent 1100 Series HPLC system equipped with a CHIRALPAK AD-H column (length 250 mm, I.D. 4.6 mm; hexanes/isopropanol 99:1, 1.0 mL/min) with t
r (minor) = 12.9 min, t
r (major) = 13.3 min. The purity (94%) was determined by GC analysis (t
r = 4.35 min), using the same conditions as for compound
1.
Safety and Waste Disposal Information All hazardous materials should be handled and disposed of in accordance with "Prudent Practices in the Laboratory"; National Academy Press; Washington, DC, 1995.
3. Discussion
The transition metal-catalyzed enantioselective coupling of allylic electrophiles with carbon nucleophiles has been the focus of intense investigation.
5 Salient examples include palladium-catalyzed couplings with enolates, nickel-catalyzed couplings with Grignard reagents, and copper-catalyzed couplings with Grignard and diorganozinc reagents.
6 Despite impressive progress, the development of methods that have broader scope with respect to the nucleophile, as well as improved functional-group compatibility, persist as important challenges.
This work describes a nickel-based catalyst for the asymmetric cross-coupling of racemic secondary allylic chlorides with readily available alkylzinc halides (Eq 1).
7,8,9 The process is stereoconvergent: both enantiomers of the substrate are transformed into the same enantiomer of the product with high stereoselectivity.
In the case of "symmetrical" allylic halides (e.g., entries 1-4 of Table 1), regioselectivity in the carbon-carbon bond formation is not an issue. As the steric demand of the substituents on the termini of the allyl fragment increases, the ee decreases (entries 1-3). A bulky 1,2,3-trisubstituted allylic chloride undergoes cross-coupling with excellent enantioselectivity, although with modest yield (entry 4). An unactivated alkyl chloride is compatible with the reaction conditions (entry 2).
Table 1. Enantioselective, Stereoconvergent Negishi Cross-Couplings of Allylic Chlorides (for the reaction conditions, see Eq 1).
In the case of unsymmetrical allylic chlorides, if the two substituents on the termini of the allyl group have similar steric and electronic properties, then the regioselectivity is modest (e.g., 1.9:1 for n-Bu vs. Me; entry 5 of Table 1). On the other hand, good regioselectivity and enantioselectivity are observed when there is substantial steric (entries 6 and 7; >20:1 regioselectivity) or electronic (entries 8-10; >20:1 regioselectivity) differentiation. Carbon-carbon bond formation occurs preferentially at the less-hindered site (entries 5-7) or at the site that preserves conjugation (entries 8 and 9).
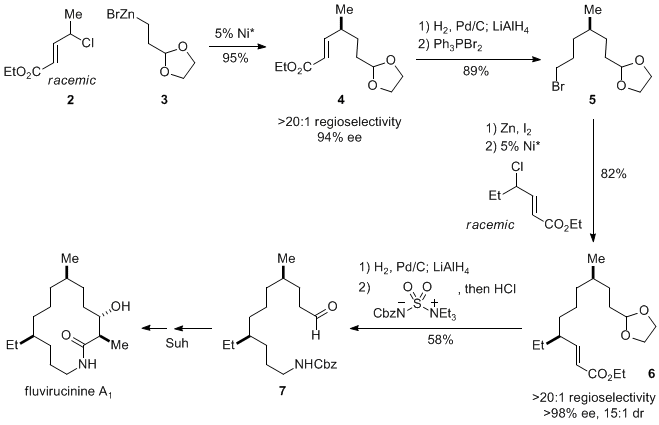
Ethyl (
E)-4-chloropent-2-enoate (
2) and 2-[2-(1,3-dioxolan-2-yl)ethyl]zinc bromide (
3) were selected as the cross-coupling partners because the product serves as an intermediate in the formal total synthesis of fluvirucinine A
1 (Scheme 1).
7,10 The Negishi reaction proceeded with a strong preference for carbon-carbon bond formation at the γ position of the electrophile, providing compound
4 in excellent yield (94-96%), regioselectivity (>20:1), and ee (94-95%). Reduction of
4, followed by bromination, furnished
5, which was converted into the organozinc reagent and subjected to a second asymmetric Negishi cross-coupling with a racemic allylic chloride to afford intermediate
6 in good yield (82%), regioselectivity (>20:1), and stereoselectivity (>98% ee; 15:1 dr). This coupling product can be transformed into aldehyde
7, which was an intermediate in Suh's total synthesis of fluvirucinine A
1.
10Scheme 1. Formal Total Synthesis of Fluvirucinine A1 via Two Catalytic Enantioselective Negishi Cross-Couplings of Allylic Chlorides ((R)-CH2CH2Ph-Pybox was used; for the reaction conditions, see Eq 1).
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
Ethyl (E)-4-oxo-2-butenoate: 2-Butenoic acid, 4-oxo-, ethyl ester, (2E)-; (2960-66-9)
Methylmagnesium bromide: Magnesium, bromomethyl-; (75-16-1)
Ethyl (E)-4-hydroxy-2-pentenoate: 2-Pentenoic acid, 4-hydroxy-, ethyl ester, (2E)-; (10150-92-2)
Dichlorotriphenylphosphorane: Phosphorane, dichlorotriphenyl-; (2526-64-9)
Ethyl (E)-4-chloropent-2-enoate: 2-Pentenoic acid, 4-chloro-, ethyl ester, (2E)-; (872455-00-0)
2-(1,3-Dioxolan-2-yl)ethyl bromide: 1,3-Dioxolane, 2-(2-bromoethyl)-; (18742-02-4)
2-[2-(1,3-Dioxolan-2-yl)ethyl]zinc bromide: Zinc, bromo[2-(1,3-dioxolan-2-yl)ethyl]-; (864501-59-7)
Nickel(II) chloride, dimethoxyethane adduct (NiCl2•glyme): Nickel, dichloro[1,2-di(methoxy-κO)ethane]-; (29046-78-4)
2,6-Bis[(S)-4,5-dihydro-4-(2-phenylethyl)-2-oxazolyl]pyridine: Pyridine, 2,6-bis[(4S)-4,5-dihydro-4-(2-phenylethyl)-2-oxazolyl]-; (1012042-02-2)
(S,E)-Ethyl 6-(1,3-dioxolan-2-yl)-4-methylhex-2-enoate: 2-Hexenoic acid, 6-(1,3-dioxolan-2-yl)-4-methyl-, ethyl ester, (2E,4S)-; (1012041 -93-8)
 |
Prof. Greg Fu was born in Galion, Ohio, in 1963. He received a B.S. degree in 1985 from MIT, where he worked in the laboratory of Prof. K. Barry Sharpless. After earning a Ph.D. from Harvard in 1991 under the guidance of Prof. David A. Evans, he spent two years as a postdoctoral fellow with Prof. Robert H. Grubbs at Caltech. In 1993, he returned to MIT, where he is currently the Firmenich Professor of Chemistry. Prof. Fu received the Springer Award in Organometallic Chemistry in 2001, the Corey Award of the American Chemical Society in 2004, and the Mukaiyama Award of the Society of Synthetic Organic Chemistry of Japan in 2006. He is a fellow of the Royal Society of Chemistry and of the American Academy of Arts and Sciences. Prof. Fu serves as an associate editor of the Journal of the American Chemical Society.His current research interests include metal-catalyzed coupling reactions and enantioselective nucleophilic catalysis. |
 |
Sha Lou was born in He Bei, China, in 1979. He received a B.S. in Chemistry from Beijing University of Chemical Technology in 2002, where he conducted undergraduate research on fullerene functionalizations with Professor Shenyi Yu. He obtained a Ph.D. degree in January 2008 from Boston University under the direction of Professor Scott E. Schaus. His graduate research focused on transition metal- and organic molecule-catalyzed asymmetric carbon-carbon bond-forming reactions and synthesis. In 2008, he joined the group of Professor Greg Fu at MIT as a postdoctoral fellow. His current research involves the development of transition metal-catalyzed enantioselective cross-coupling reactions. |
 |
David I. Chai was born in Korea in 1981. He obtained a B.S. in Chemistry in 2006 from the University of British Columbia while working in the laboratory of Professor Marco Ciufolini. He is currently a chemistry graduate student with Professor Mark Lautens, working on palladium-catalyzed carbo- and hetero-cycle synthesis. |
 |
Gavin Chit Tsui was born in China and raised in Hong Kong. He obtained his Hon. B. Sc. (2004) and M. Sc. (2006, supervisor: Prof. William Tam) degrees from the University of Guelph, Ontario, Canada. He then joined pharmaceutical companies (NPS Pharmaceuticals, Inc. and Merck Frosst Canada) as a research chemist in the medicinal chemistry division. Currently he is pursuing a Ph. D. degree at the University of Toronto under the supervision of Prof. Mark Lautens to investigate novel rhodium-catalyzed reactions. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved