Org. Synth. 2020, 97, 38-53
DOI: 10.15227/orgsyn.097.0038
Preparation of (-)-Levoglucosenone from Cellulose Using Sulfuric Acid in Polyethylene Glycol
Submitted by J. Klepp,
# W. Dillon,
# Y. Lin,
% P. Feng,
& and B. W. Greatrex
#1*
Checked by Yusuke Imamura, Masanori Nagatomo, and Masayuki Inoue
1. Procedure (Note 1)
A.
(-)-Levoglucosenone (LGO, 1). Polyethylene glycol 4000 (PEG) (200 g) (
Note 2) is ground to a course powder (Figure 1) in a mortar (φ: 200 mm) and pestle (203 mm) and placed in a 500 mL beaker. A diluted solution of
sulfuric acid (2.50 g, 25.5 mmol) in
methanol (15 mL) (
Note 3) is added at ambient temperature (22 °C) and the powder is mixed using a large spatula (18 cm) to distribute the acid into the powdered PEG and allowing most of the
methanol to evaporate. Cellulose powder (50 g, 308 mmol of monomer) (
Note 4) is added and the powders combined by stirring with the spatula for three minutes giving an intimate mixture of PEG/acid and cellulose powders (Figure 2). The powder mixture is transferred using a large powder funnel into a 1 L three-necked, round-bottomed flask. The left neck is equipped with a distillation head with the water-cooled Liebig condenser and 100 mL receiver flask. The center neck is equipped with a 29/32 cap, and the right neck is equipped with a thermocouple inserted through a 15/25 rubber septum (Figure 3). Two traps are connected to distillation setup. The first one is 100 mL two-necked flask and the second trap is a cold-finger. Each trap is cooled to -78 °C using a dry ice/
methanol bath.
Figure 1. Ground PEG 4000
Figure 2. Diluted H2SO4 being added to PEG (left) and appearance of the PEG/acid/cellulose mixture (right)
Figure 3. Pyrolysis setup (top), and cold traps (bottom left: 1st trap, bottom right: 2nd trap).
The setup is evacuated using a rotary-vane oil pump to less than 1.0 mmHg and the mixture heated over 15 min to 170 °C by mantle heater (
Note 5). During this time the solid PEG/cellulose mixture melts, the colorless solution begins to char and turn black. When the solution reaches 170 °C the pressure is 3 mmHg, formation of a pale yellow distillate is observed and the distillate begins to collect in the receiver flask. A heat gun is used to warm the distillation head to assist distillation of the LGO and to avoid reflux of the mixture. Heating is continued and the temperature is kept between 180-190 °C for 30 min at which point no more product distills (
Note 6). Upon completion of the distillation, a pale yellow liquid (29.8-31.0 g) is obtained, which contains mainly LGO, furfural and water (Figure 4) (
Note 7).
Figure 4. Appearance of the PEG and charred cellulose (left) and the distillate obtained (right)
The entire procedure is repeated and the distillates from the two independent reactions are combined. In total, 100 g of cellulose is pyrolyzed (
Note 8). Ethyl acetate (60 mL) (
Note 9) is added to the distillate at ambient temperature (22 °C) followed by solid
NaHCO3 (5 g) (
Note 10) and a 3.1 cm Teflon-coated magnetic oval stir bar. The reaction is allowed to stir for 15 min to facilitate evolution of CO
2 gas. The mixture is transferred to a 100 mL separating funnel and the flask is washed with
ethyl acetate (5 mL), which is added to the separatory funnel. The organic phase is separated and the aqueous phase is extracted with an additional portion of
ethyl acetate (60 mL) (Figure 5) (Notes
9 and
11). The combined organic layers are dried (
Na2SO4, 3.0 g) (
Note 12), then filtered through a filter funnel fitted with a plug of cotton wool into a 500 mL round-bottomed flask. The volatiles are removed using a rotary evaporator (37 °C, 20 mmHg) to give a yellow-orange oil (28 g) containing mainly furfural and LGO (
Note 13).
Figure 5. Extraction 1 (E1, left), extraction 2 (E2, middle) and TLC (right) of the extractions 1-3, residual aqueous layer and reference (KMnO4 stain)
The residue is purified by flash column chromatography on silica (
Note 14) using 10:1 hexane/
ethyl acetate (Notes
9,
15, and
16). Purification is also possible through distillation (Notes
17 and
18). The product received from chromatographic purification (Figure 6) is concentrated on the rotary evaporator (37 °C, 20 mmHg) and afforded LGO as a yellow oil (13.9 g, 18%, 97.5% purity by qNMR) (Notes
19,
20 and
21).
Figure 6. TLC (Note 21) of column fractions F16-F40 (KMnO4 stain, left) and isolated LGO (right)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with polyethylene glycol 4000 (PEG),
sulfuric acid,
methanol, cellulose powder, dry ice,
acetone,
ethyl acetate,
sodium bicarbonate, hexanes,
sodium sulfate, silica,
chloroform, and
chloroform-d.
2. PEG 4000 was purchased from Sigma-Aldrich. Although PEG 4000 was used in this reaction, longer chain polymers including PEG 6000 (Merck) gave equivalent results. The use of PEG 400 gave lower yields of product and a range of cyclic crown ethers were observed by GCMS which were presumably formed when PEG 400 underwent acid-catalyzed depolymerization. These crown ethers were not observed in PEG 4000 reactions as long as temperatures were kept below 205 °C.
3.
Sulfuric acid (95-98%) and
methanol (ACS reagent) were purchased from Sigma-Aldrich and used as received.
4. Standard grade Whatman ashless cellulose powder was used (submitters). Microcrystalline cellulose (Avicel, Sigma-Aldrich) was used as received (checkers).
5. Since this pyrolysis reaction happens at high temperature, it is critical to ensure the operation is within a thermo-safe zone. As such, differential scanning calorimetry (DSC) analysis was performed on the reaction mixture and products (Figure 7a and 7b). DSC curves from a sealed reaction mixture indicated a rapid exotherm (1.02 W/g, about 10 °C increase/min, the safe zone is below 0.33 W/g, or 3 °C increase/min) starting at 174 °C and ending at 234 °C. Subsequent DSC analysis of the individual components PEG 4000, cellulose, and reaction residual mixture indicated negative exotherm risk from 160 °C to 220 °C. The yellowish crude product exhibits a rapid exotherm (2.28 W/g) in the DSC curve from 134 °C to 227 °C. The data indicated that the exotherm observed in the reaction mixture derived from the crude product mixture from the reaction. Therefore, the key to avoid any safety risk is the effective and rapid removal of the crude product under vacuum, and therefore an efficient vacuum pump is necessary for this procedure.
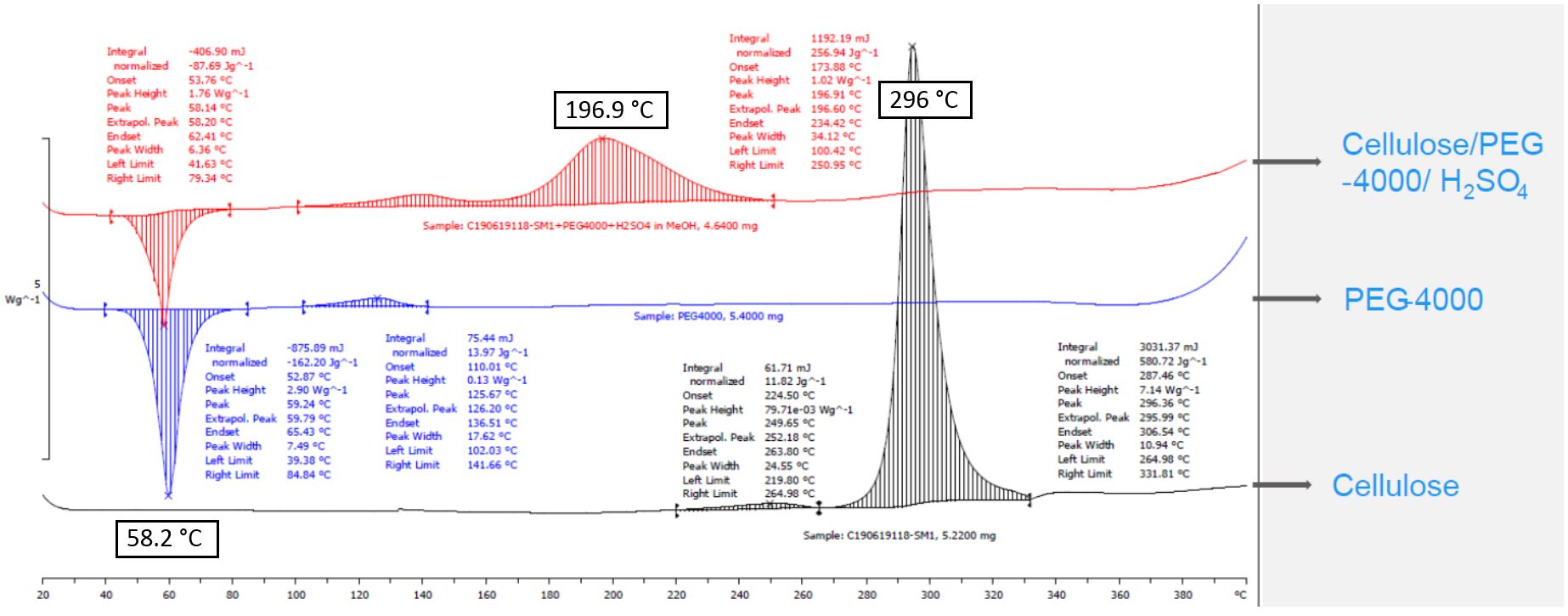
Figure 7a. DSC of reaction mixture, PEG 4000, and cellulose
Figure 7b. DSC of crude product and undistilled reaction residue
6. Pressure plays a role in the outcome of this reaction. Relatively low yields of LGO were using a two-stage diaphragm pump for vacuum compared to the higher vacuum generated with a rotary vane oil pump. LGO has a boiling point of 113 °C at 3 mmHg. Higher vacuums improve yields as they allow rapid removal of the product from the acidic mixture.
7. The proportion of water, LGO and furfural is dependent on the vacuum in the experimental setup. Stronger vacuums result in less water and furfural condensed in the primary distillate with more water collecting in the cold fingers. Furfural is also removed if the reaction mixture is kept under high vacuum for extended periods of time.
8. Performing the reaction with a single batch of 100 g cellulose results in a reduction in the yield due to excessive LGO decomposition during pyrolysis. The PEG and char can be recycled after cooling. Replacing fresh PEG in the procedure with the recycled PEG/char mixture results in a 27% reduction in LGO yield.
9. EtOAc (99.0%) was purchased from Kanto Chemical Co. Ltd. and was used as received.
10.
NaHCO3 (>99.5%) was purchased from Nacalai Tesque, Inc. and used as received.
11. The presence of LGO in the extracts is monitored using TLC (1:4
ethyl acetate/hexanes) (
Note 22). A third extraction is found to contain virtually no additional LGO.
12. Anhydrous
Na2SO4 (99%) was purchased from FUJIFILM Wako Pure Chemical Corporation and used as received.
13.
1H NMR
pdf analysis of the crude reaction product indicated a mixture of mainly furfural and LGO.
14. Silica gel 60 (0.040-0.063 mm) was purchased from Merck KGaA and was used as received. Silica gel 60 (200 g) is loaded into a 5.5 cm diameter x 35 cm height column using 10:1 hexane/
ethyl acetate.
15. Hexane (95.0%) was purchased from Kanto Chemical Co. Ltd. and used as received.
16. The material obtained from the extraction is mixed with 10:1 hexane/
ethyl acetate (20 mL) and loaded onto the silica gel. A gradient of hexane/
ethyl acetate (20 x 200 mL, 10:1 hexane/
ethyl acetate followed by 20 x 200 mL, 5:1 hexane/
ethyl acetate) is eluted. LGO is obtained in fractions 22 to 36 (Figure 6).
17. For the amount of product produced on this scale, flash chromatography was found to be more reliable. Purification of the residue by distillation under vacuum is also possible; however, complete separation of lower boiling furfural from the LGO requires a long fractionating column and decomposition of the LGO can occur if acid remains in the mixture. Vacuum distillation (0.5 mmHg) of the furfural/LGO mixture before chromatography, from a 25 mL tear-shaped flask fitted with an 11 cm Vigreux column (Figure 8), gave two fractions. The fraction boiling at 63-81 °C contained LGO (1.47 g, 1.5%, 80% purity) contaminated with furfural and the fraction boiling at 81-86 °C contained LGO as a pale yellow oil (6.83 g, 8.9%, 97% purity). Purity assessment was performed using by Q NMR using dimethylfumarate as an internal standard.
Figure 8. Alternate purification of the final product by distillation under vacuum
18. The initial pH of the distillate was 1.65. After addition of
NaHCO3 the pH was ~6.0 measured using a glass electrode pH meter. If this step was omitted, a polymeric material can form upon concentration. The addition of salts to the water/LGO/furfural mixture allows for the extraction of LGO from the mixture, which is otherwise difficult. At higher pH values, hydration of the LGO can occur; however, this is not observed with the method described.
19.
(-)-Levoglucosenone (LGO, 1) has the following properties: [α]
D22 -523 (
c = 1.0,
CHCl3); Lit
2 [α]
D31 -515 (
c = 0.3,
CHCl3);
1H NMR
pdf (400 MHz, CDCl
3) δ: 3.76 (br d,
J = 6.8 Hz, 1H), 3.89 (dd,
J = 7.3, 4.5 Hz, 1H), 5.00 (dd,
J = 4.8, 4.8 Hz, 1H), 5.35 (d,
J = 1.4 Hz, 1H), 6.11 (dd,
J = 10.0, 1.4 Hz, 1H), 7.28 (dd,
J = 10.0, 4.5 Hz, 1H);
13C NMR
pdf (100 MHz, CDCl
3) δ: 66.6, 71.7, 101.6, 126.9, 148.0, 188.8; IR (neat) 2968, 2899, 1715, 1697, 1378, 1255, 1106 cm
-1. Purity of 97.5% was assessed by qNMR
pdf using dimethyl fumarate as the internal standard. The product shows no evidence of decomposition when stored at 4 °C for more than a year.
20. When the reaction was carried out on half scale, 5.87 g (15%) of the product was obtained.
21. The preparation procedure was also repeated by chemists from STA Pharmaceutical Co., Ltd. (STA) and MSD, similar yields (11-13%) were obtained.
22. TLC Silica gel 60 F
254 was purchased from Merck KGaA and used as received (checkers).
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Levoglucosenone (LGO) is a small chiral molecule formed when cellulose or levoglucosan is heated in the presence of acid.
3 It is being developed as a biorenewable platform chemical that can be produced without costly fermentation following acid pretreatment of lignocellulosic biomass, although the procedure described here uses readily available cellulose powder in place of biomass.
LGO has been prepared from cellulose using a number of catalysts including using
sulfuric acid3,4 and phosphoric acid in sulfolane,
4 and from lignocellulosic sources including acid-treated poplar wood
5 and sugar cane bagasse.
6 Previous syntheses performed in the absence of solvent have reported the formation of LGO at higher temperatures than those reported here. For example, Dobele reported conversion of cellulose to LGO started at 240 °C with maximum conversion of 22.3% obtained at 350 °C on a 100 mg scale.
7 In the presence of solvent, the temperatures required for the formation of LGO are much lower. For example, Kawamoto's study which used only 50 mg of cellulose in sulfolane with 5%
sulfuric acid generated up to 42% LGO at 240-280 °C.
4 Recently Huber et al. reported the conversion of cellulose into LGO using THF and
sulfuric acid at the relatively low temperature of 190 °C, however, the method required a high-pressure reactor and loadings of greater than 1% cellulose gave reduced yield.
8 Recently, DuPont Chemicals has patented a process to make LGO from biomass which lists PEG and
sulfuric acid in one of the embodiments, however, the loading of cellulose in the described procedure was only 5% using 2.5-3.0 g of biomass and precise procedures were not provided.
9 Importantly, most previous studies made no attempt to purify the LGO and report conversions in crude pyrolysates and not isolated yields. Furthermore, early attempts at scaling the synthesis of LGO have met with failure, the compound is still expensive (> $200/g) which may indicate that these procedures do not have scalability past milligram quantities. Preparative scale procedures have been reported by Marshall (330 mg scale, 9.6% yield) who performed cellulose pyrolysis in soybean oil and Trahanovsky et. al. (200-300 mg, 5% yield), who used unspecified vegetable oil.
10,11 Isobe et al. described the solvent-free production of 104 g of LGO from 5.8 kg of acidified cellulose at 300-350 °C equating to a 1.8% yield.
12 We have found that the use of PEG 4000 lowers the temperature at which LGO forms relative to studies which have omitted solvent, and as it does not distill, separation is simplified compared to reactions carried out in sulfolane. The formation of LGO at lower temperatures may be rationalized by the stabilization of the oxonium and oxocarbenium ions along the pathway of its formation.
13LGO can be used to synthesize biologically active molecules and in particular is a useful synthon for the production of γ-lactones.
14,15 Targets accessible from LGO include tetrodotoxin,
16 eldanolide,
17 PCCG-IV,
18 and the antiviral drug indinavir.
19,20 LGO can also be used as a scaffold for the development of chiral organocatalysts, and highly selective chiral auxiliaries.
21,22,23 Furthermore, the dihydro-derivative (Cyrene)
has been proposed as a biorenewable solvent as many parameters are similar to N-methylpyrrolidine and DMF and abundant lignocellulose can be used as a precursor material.
24
Appendix
Chemical Abstracts Nomenclature (Registry Number)
(LGO) (1): Levoglucosenone; (37112-31-5)
PEG 4000: Poly(ethylene glycol); (25322-68-3)
Sulfuric acid; (7664-93-9)
Cellulose; (9004-34-6)

|
Julian Klepp received his M.Sc. degree from the University of Strasbourg, France and University of Stuttgart, Germany under the supervision of Professor René Peters in 2016. He moved to Armidale, Australia to pursue his doctoral studies under the supervision of Professor Ben Greatrex. His initial graduate studies were aimed towards the development of asymmetric catalysts and his current research is focused on exploiting the potential of biomass as a source of chiral building blocks.
|

|
Wayne Dillon received a First Class Honours Degree (BSc Chemistry and Physics) from the University of New England, Australia in 2009. After working as a research assistant and instrument technician he recently began a Ph.D. at the University of Otago, New Zealand under the supervision of Dr Christina McGraw and Professor Cliff Law. His doctoral research is aimed at the development of deployable sensing devices for marine and agricultural applications.
|

|
Yipeng Lin received his B.S. from the North University of China in 2008. He then joined Shanghai Institute of Materia Medica (SIMM), Chinese Academy of Sciences (CAS) as a chemical engineer, where his research mainly focused on discovery of novel Androgen Receptor antagonist for the treatment of prostate cancer under supervision of Professor Wenhu Duan. Since 2016, He joined WuXi AppTec and served as a senior scientist to devote himself to pharmaceutical process research & development.
|

|
Feng Peng joined the Process Research Department of Merck & Co., Inc. in 2012. His research focuses on using state-of-art organic chemistry to address critical problems in drug development. He received his B. S. degree from Beijing Normal University. He obtained his M.S. under the supervision of Professor Dennis Hall at University of Alberta with a research focus on Boron Chemistry. Feng then moved to New York City, where he obtained Ph.D. in the area of total synthesis (maoecrystal V) with Professor Samuel Danishefsky at Columbia University.
|

|
Ben Greatrex completed his Ph.D. in 2004 at the University of Adelaide, Australia with Professor Dennis Taylor before moving to New Zealand for post-doctoral study with Prof. Richard Furneaux. He began his independent research career in 2010 at the University of New England, Armidale. His research interests include synthetic methodology, reaction mechanisms and natural products chemistry.
|

|
Yusuke Imamura was born in New York, USA. He graduated from the University of Tokyo in 2017 with B.S. in Pharmaceutical Sciences. He is continuing his graduate studies at the University of Tokyo under the supervision of Prof. Masayuki Inoue. His research interests are in the area of the total synthesis of complex natural products.
|

|
Masanori Nagatomo completed his Ph.D. in 2012 from the University of Tokyo under the supervision of Professor Masayuki Inoue. In 2012, he carried out visiting research with Professor Phil S. Baran at The Scripps Research Institute. In the same year, he was appointed as Assistant Professor in the Graduate School of Pharmaceutical Sciences at the University of Tokyo and promoted to Lecturer in 2018. His research efforts focus on the development of novel synthetic methodology and applications to the multistep synthesis of complex molecules.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved