Org. Synth. 2020, 98, 374-390
DOI: 10.15227/orgsyn.098.0374
Synthesis and Acylation of 1,3-Thiazinane-2-thione
Stuart C. D. Kennington, Oriol Galeote, Miguel Mellado-Hidalgo, Pedro Romea,
1* and Fèlix Urpí
1*
Checked by Zhaobin Han and Kuiling Ding
1. Procedure (Note 1)
A. 3-Ammoniopropylsulfate (1). An oven-dried single-necked 100 mL round-bottomed flask (14/23 joint), equipped with a 2.5-cm Teflon-coated magnetic stirbar, is charged with 3-amino-1-propanol (11.5 mL, 150 mmol, 1 equiv) (Note 2) and anhydrous dichloromethane (35 mL) (Note 3). A 50 mL pressure-equalizing addition funnel (14/23 joint) equipped with a CaCl2 tube is attached to the round-bottomed flask and is then charged with chlorosulfonic acid (10.5 mL, 159 mmol, 1.06 equiv) (Note 4) using a 20 mL glass luer-lock syringe. The flask is immersed in an ice/water bath and the solution is stirred for 5 min. The chlorosulfonic acid is added dropwise over 30 min, allowing the fumes to escape. A white precipitate is formed during the addition. Once the addition is complete, the reaction is stirred at 0 °C for 20 min and left to warm slowly to room temperature over 30 min (Note 5). Once at room temperature, the reaction mixture is stirred for 1 h. The resulting mixture is filtered through a 70 mm diameter Number 3 Glass filter funnel with a Büchner setup. A bent spatula and methanol (25 mL) (Note 6) are used to remove remaining product from the flask walls. The mixture in the filter funnel is triturated with methanol (40 mL, then 2 × 20 mL) (Note 6), using a spatula to break up the lumps each time. The resulting white solid is broken up into a coarse powder and transferred to a 100 mL round-bottomed flask (29/32 joint), where it is placed on a rotary evaporator (40 °C, 12 mmHg pressure) for 1 h. The resulting white solid is ground to a fine white powder using a 10 cm diameterglass mortar and pestle, retransferred to a 100 mL round-bottomedflask and dried on a high vacuum line (25 °C, 0.1 mmHg pressure) for 2 h giving the title compound 1 (20.72 g, 134 mmol, 89% yield) (Note 7) as a fine white powder.

Figure 1. Addition set-up (left), trituration (middle) and final product (right)
(photos provided by submitters)
B.1,3-Thiazinane-2-thione (2). An oven-dried single-necked 250 mL round-bottomed flask (29/32 joint), equipped with a 4-cm Teflon-coated magnetic stirbar, is charged with 3-ammoniopropylsulfate (1) (18.70 g, 121 mmol, 1 equiv) and absolute ethanol (15 mL) (Note 8) at room temperature. The resulting solution is stirred at 25 °C for 3 min and neat carbon disulfide (9.6 mL, 160 mmol, 1.3 equiv) (Note 9) is added in one portionusing a 10 mL syringe.
Separately, KOH beads (14.84 g, 265 mmol, 2.2 equiv) (Note 10) are weighed in a 250 mL conical flask equipped with a4-cm Teflon-coated magnetic stirbar, and the KOH is dissolved in 1:1 ethanol/water (100 mL). The mixture is stirred at 25 °C, and the resulting solution is transferred with a funnel to a 250 mL pressure-equalizing addition funnel (29/32 joint) attached to the round-bottomed flask. The neck of the addition funnel is sealed with a rubber septum, the system is purged with a nitrogen flow for 5 min, and a nitrogen atmosphere is maintained via a needle inserted into the septum. The KOH solution is added dropwise to the carbon disulfide solution in the round-bottomed flask over 30 min at room temperature to give a yellow solution. The addition funnel is replaced by a reflux condenser sealed with a rubber septum, and a nitrogen is provided by a needle inserted through the septum. The reaction mixture is heated to reflux in an aluminum heating block (70 °C) for 1 h under a N2 atmosphere and allowed to cool to room temperature slowly to give a fluffy white precipitate in the solution, which is further cooled to 0 °C with an ice/water bath for 15 min.
The mixture is filtered using a 70 mm diameter Number 3 glass filter funnel with a Büchner setup. The flask is rinsed with cold deionized water (3 × 15 mL) with the washings being added to the filter funnel. The solid in the filter funnel is dried under vacuum (20 mmHg)for 15 min, after which the receiving flask is changed for a fresh one. The solid is washed with dichloromethane (3 × 35 mL) (Note 11), each time breaking up the solid with a spatula and mixing thoroughly before applying the vacuum. The combined organic extracts are dried over MgSO4 (40 g) (Note 12) and concentrated under reduced pressure (12 mmHg) to give pure crystalline powder of 1,3-thiazinane-2-thione 2 (6.78 g, 42% yield) (Note 13). The remaining solid (Note 14) from the filter funnel is transferred to a 250 mL round-bottomed flask (29/32 joint) equipped with a 4-cm Teflon-coated magnetic stir bar. This round-bottomed flask is charged with dichloromethane (150 mL) (Note 11), a reflux condenser sealed with a rubber septum is attached to the flask, the system is purged with a nitrogen flow for a couple of minutes, anda nitrogen atmosphere is maintained through the septum.The resulting mixture is stirred and heated to reflux for 1 h, and the warm solution is filtered through a Number 3 glass filter funnel with a Büchner setup. The solid is washed with dichloromethane (2 × 50 mL) (Note 11), each time breaking up the solid with a spatula and mixing thoroughly before applying the vacuum (20 mmHg) as before. The combined filtrates are dried over MgSO4 (35 g) (Note 12) and concentrated (12 mmHg) to give pure thiazinanethione 2 (3.34 g, 21% yield). The combined thiazinanethione 2 weighs 10.12 g (63% overall yield) (Note 15).

Figure 2. Reaction set-up (left), reflux set-up (middle) and final product (right)
(photos provided by submitters)
C. N-Propanoyl-1,3-thiazinane-2-thione (3). An oven-dried single-necked 250 mL round-bottomed flask (29/32 joint), equipped with a 4-cm Teflon-coated magnetic stirbar, is charged with 1,3-thiazinane-2-thione (10.64 g, 80 mmol, 1 equiv). The flask is sealed with a rubber septum and the system is flushed with a flow of nitrogen. The flask is charged with anhydrous dichloromethane (80 mL) (Note 3) via syringe and immersed in an ice/water bath. The resulting solution is stirred for 2 min and freshly distilled triethylamine (14.5 mL, 104 mmol, 1.3 equiv) (Note 16) is added dropwise over 2 min. The solution is stirred for 2 min and propionyl chloride (8.4 mL, 96 mmol, 1.2 equiv) (Note 17) is carefully added over 20 min, which produces a yellow solution. The ice/water bath is removed,and the reaction mixture is allowed to warm to room temperature for 2 h. The resulting dark yellow/orange mixture is cooled with an ice/water bath, quenched with the addition of a saturated solution of NH4Cl (25 mL) (Note 18) in one portion, and allowed to stir for 5 min.
The mixture is transferred to a 500 mL separating funnel. The flask is rinsed with dichloromethane (4 × 40 mL) (Note 11) and water (3 × 40 mL), which are added to the separating funnel. The mixture is shaken vigorously and the layers separated. The aqueous layer is extracted with another portion of dichloromethane (40 mL) (Note 11). The combined organic extracts are washed with 2 M NaOH (120 mL) (Note 19), dried over MgSO4 (25 g), and filtered, with the flask and solid being rinsed with dichloromethane (3 × 40 mL) (Note 11), which is added to the filtrate. The solution is concentrated under reduced pressure (12 mmHg) and the resulting residue is submitted to column chromatography on silica gel (60 Å) using hexanes (Note 21) and ethyl acetate (Note 22) as eluent. Chromatographic purification (Note 23) provided pure acylated product 3 (12.74 g, 84% yield) (Note 24) (Figure 3).
Figure 3. Appearance of product 3 (photo provided by submitters)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
chlorosulfonic acid,
carbon disulfide,
triethylamine,
propionyl chloride,
sodium hydroxide,
potassium hydroxide,
Celite®, silica gel,
dichloromethane,
methanol,
hexane, and
ethyl acetate.
2.
3-Amino-1-propanol (99%) was purchased from Acros Organics and used as received.The checkers purchased
3-Amino-1-propanol (>99%) from Tokyo Chemical Industry Co., Ltd. and was used as received.
3.
Dichloromethane (99%) was purchased from Acros Organicsand was freshly distilled from CaH
2. The checkers purchased
dichloromethane (99%) from Sinopharm Chemical Reagent Co., Ltd. and the solvent was freshly distilled from CaH
2.
4.
Chlorosulfonic acid (99%) was purchased from Acros Organics and used as received. The checkers purchased
chlorosulfonic acid (>97%) from Tokyo Chemical Industry Co., Ltd. and used the reagent as received.
5. If the reaction becomes too vigorous it can be kept cooled.
6.
Methanol (99%) was purchased from Acros Organicsand used as received. The checkers purchased
methanol (99%) from Sinopharm Chemical Reagent Co., Ltd. and used the solvent as received.
7.
3-Ammoniopropylsulfate (
1) has the following physical and spectroscopic properties: mp 205-207 °C; IR (film): 3126, 3064, 2976, 1627, 1530, 1195, 1169, 1065, 923, 759, 574 cm
-1;
1H NMR
pdf (400 MHz, DMSO-d
6) δ: 1.74-1.86 (m, 2H), 2.76-2.90 (m, 2H), 3.81 (t,
J = 6.0 Hz, 2H), 7.64 (br s, 3H);
13C NMR
pdf (100.6 MHz, DMSO-d
6) δ: 27.3, 36.7, 63.2; HRMS (+ESI)
m/z calcdfor C
3H
10NO
4S [M + H]
+ 156.0325, found 156.0322.The purity of
1 was determined to be 97% by
1H qNMR
pdf using 10.8 mg of ethylene carbonate(>99% purity) as an internal standard and 18.7 mg of compound
1. Based on this purity, the actual amount of
1 formed in the reaction was 20.1 g. A second reaction on identical scale provided 20.84 g (89%, uncorrected for purity) of the product
1.
8. Absolute ethanol (99%) was purchased from Acros Organicsand used as received.The checkers purchasedabsolute ethanol (99%) from Sinopharm Chemical Reagent Co., Ltd. and used the solvent as received.
9.
Carbon disulfide was purchased from Sigma Aldrich (ACS reagent, 99.9%) and used as received.The checkers purchased
carbon disulfide (>98%) from Tokyo Chemical Industry Co., Ltd. and used it as received.
10.
KOH (98%) beads was purchased from Panreac and used as received.The checkers purchased
KOH (99%) from Sinopharm Chemical Reagent Co., Ltd. and used it as received.
11.
Dichloromethane (99%) was purchased from Acros Organicsand used as received.The checkers purchased
dichloromethane (99%) from Sinopharm Chemical Reagent Co., Ltd. and used the solvent as received.
12. Anhydrous MgSO
4 was purchased from Panreac and used as received.The checkers purchasedanhydrous MgSO
4 (99%) from Sinopharm Chemical Reagent Co., Ltd. and used the drying agent as received.
13.
1,3-Thiazinane-2-thione (
2) has the following physical and spectroscopic properties: mp 138-140 °C [lit.
2 mp 132-133 °C]; IR (film,cm
-1): 3140, 3049, 3000, 2918, 2848, 1543, 1426, 1353, 1331, 1273, 1182, 1084, 1011, 897, 748, 625;
1H NMR
pdf (400 MHz, CDCl
3) δ: 2.13-2.23 (m, 2H), 3.00 (t,
J = 6.0 Hz, 2H),3.48 (t,
J = 5.6 Hz, 2H),8.75 (br s, 1H);
13C NMR
pdf (100.6 MHz, CDCl
3) δ: 20.5, 30.1, 44.3,194.5; HRMS (+ESI)
m/z calcdfor C
4H
8NS
2 [M + H]
+ 134.0093, found 134.0091.The purity of
2 was determined to be 99% by
1H qNMR
pdf using 16.4 mg of 1,3,5-dimethoxybenzene (>99% purity) as an internal standard and 14.3 mg of compound
2.
14. The remaining solid is a thick paste containing impurities formed in the reaction.
15. The quantities of bothcrops can vary, but the overall yield and the characterization data for the two crops are consistent.A second reaction on identical scale provided 10.06 g (62%) of the product
2.
16.
Triethylamine (99%) was purchased from Fluorochemand was freshly distilled over CaH
2.The checkers purchased
triethylamine (99%) from Sinopharm Chemical Reagent Co., Ltd. and it was freshly distilled over CaH
2.
17.
Propionyl chloride (99%) was purchased from Acros Organics and used as received.The checkers purchased
propionyl chloride (>98%) from Tokyo Chemical Industry Co., Ltd. and it was used as received.
18. Ammonium chloride was purchased from Panreac and used as received.The checkers purchasedammonium chloride (99%) from Sinopharm Chemical Reagent Co., Ltd. and it was used as received.
19.
NaOH (98%) beads were purchased from Panreac and they were used as received.The checkers purchased
NaOH (99%) from Sinopharm Chemical Reagent Co., Ltd. and used it as received.
20. Silica gel was purchased from Sigma Aldrich.The checkers purchased silica gel (SiliaFlash P60, particle size: 40-63µm, pore size 60Å) from SILICYCLE.
21. Hexanes (99%) was purchased from VWR Internationaland used as received.The checkers purchased hexanes (99%) from Tansoole.
22.
Ethyl acetate was purchased from Panreac and used as received.The checkers purchased
ethyl acetate (99%) from Tansoole.
23. A 6 cm diameter column with a length of 25 cm contained silica (
ca 400 g) (
Note 20). The silica is first compacted with 90:10 hexanes/
ethyl acetate (1 L) (Notes
21 and
22) and the surface levelled. The crude residue is dissolved in
ethyl acetate (4 mL) (
Note 22), diluted in hexanes (8 mL) (
Note 21), and added onto the compacted column. After adsorption, the flask that contained the crude residue is washed with 90:10 hexanes/
ethyl acetate (3 × 8 mL, or until the flask is no longer yellow), each time waiting until the liquid is adsorbed. The walls of the column are then washed with 90:10 hexanes/
ethyl acetate (3 × 8 mL) (Notes
21 and
22). Once all of the yellow product is adsorbed on the silica, a thick layer of sand is added to protect the silica. The column is eluted with 90:10 hexanes/
ethyl acetate (
ca 4.5 L) until all of the yellow color (Figure 4) has left the column and the eluent runs clear. The product is collected in
circa 15030 mL-test tubes. Contents of the tubes are assessed by TLC (80:20 hexanes/
ethyl acetate; R
f = 0.39) (Figure 5), and those containing pure product are sequentially added to a 1 L round-bottomed flask and concentrated (20 mmHg, 40 °C). Once the product is concentrated, the resulting thick yellow oil is diluted with
dichloromethane (50 mL) (
Note 11), and the solution transferred to a 100 mL round-bottomed flask. The solution is concentrated under reduced pressure (20 mmHg) and kept under high vacuum (0.1 mmHg) at room temperature for 4 h to afford the product (
3).
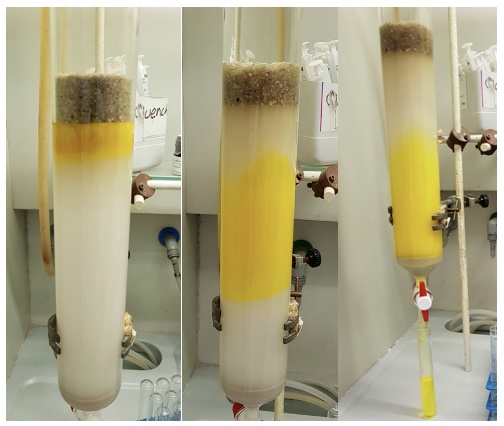
Figure 4. Product 3 (yellow) eluting down the silica column
(photos provided by submitters)
Figure 5. Image of a TLC plate (UV) of a pure sample
(photos provided by checkers)
24.
N-Propanoyl-1,3-thiazinane-2-thione (
3) has the following physical and spectroscopic properties: yellow-orange oil; R
f 0.39 (80:20 hexanes/
EtOAc); IR (film): 2975, 2934, 2877, 1700, 1469, 1374, 1344, 1302, 1198, 1166, 1020, 966, 919 cm
-1;
1H NMR
pdf (400 MHz, CDCl
3) δ: 1.22 (t,
J = 7.2 Hz, 3H), 2.19-2.30 (m, 2H),3.04 (t,
J = 6.8 Hz, 2H),3.09 (q,
J = 7.2 Hz, 2H), 3.91-3.95 (m, 2H);
13C NMR
pdf (100.6 MHz, CDCl
3) δ: 10.1, 22.9, 32.1, 32.7, 46.6, 179.0,202.9; HRMS (+ESI)
m/z calcdfor C
7H
12NOS
2 [M + H]
+ 190.0355, found 190.0354.The purity of
3 was determined to be >98.5% by
1H qNMR
pdf using 16.8 mg of 1,3,5-dimethoxybenzene (>99% purity) as an internal standard and 22.6 mg of compound
3. Based on this purity, the actual amount of
3 formed in the reaction was 12.57 g. A second reaction on identical scale provided 12.68 g (84%, uncorrected for purity) of the product
3.
Working with Hazardous Chemicals
The procedures in
Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red "Caution Notes" within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
Chiral heterocycles are widespread platforms from which a variety of stereoselective transformations can be carried out.
3 Indeed, chiral oxazolidinones (Scheme 1) introduced by Evans in the 80s still hold a prominent position among the most efficient chiral auxiliaries
4,5,6 and are currently employed for the synthesis of natural products.
7 Furthermore, structurally similar oxazolidinethiones and thiazolidinethiones (Scheme 1) have also played a crucial role in stereoselective synthesis.
8,9,10,11 Irrespective of the high stereocontrol provided by such auxiliaries, their prevalence is also due to their straightforward synthesis from proteinogenic α-amino acids (Scheme 1).
5,6,8,12Scheme 1. Heterocycles used as chiral auxiliaries
Importantly, the quest for new catalytic and asymmetric transformations adhered to the atom economy principle
13 have also aroused the interest in the achiral counterparts of such heterocycles. Indeed, Evans reported the crucial role played by achiral
N-Propanoyl-1,3-thiazinane-2-thione in enantioselective aldol reactions catalyzed by a chiral nickel(II) complex (Scheme 2).
14Scheme 2. Achiral N-propanoyl-1,3-thiazolidine-2-thione in enantioselective aldol reaction
Five membered achiral scaffolds may thus mimic the performance of their chiral counterparts shown in Scheme 1. Surprisingly, there is a lack of similar reactions based in the corresponding six membered heterocycles.
2 In this context, we have recently reported that
N-Propanoyl-1,3-thiazinane-2-thioneundergoes highly efficient alkylation reactions with benzhydryl
Scheme 3. Alkylation of N-propanoyl heterocycles with (4-MeOC6H4)2CHOMe
methyl ethers, slightly more enantioselective than the parallel thiazolidinethione scaffold (Scheme 3).
15 Therefore, a six-membered thiazinane heterocycle may be a valuable platform for asymmetric synthesis.
Acylation of 1,3-thiazinane-2-thione with acyl chlorides at 10-15 mmol provides the corresponding N-acyl thiazianethiones with yields up to 93% (Scheme 4. Part A). Alternatively, such acylations can be carried out by coupling of 1,3-thiazinane-2-thione and carboxylic acids with EDC at 5-10 mmol scale (Scheme 4. Part B).
Scheme 4. Acylation of 1,3-thiazinane-2-thione
Appendix
Chemical Abstracts Nomenclature (Registry Number)
3-Amino-1-propanol: 1-Propanol, 3-amino-; (156-87-6)
Chlorosulfonic acid: Chlorosulfuric acid; (7790-94-5)
3-Ammoniopropylsulfate: 1-Propanol, 3-amino-, 1-(hydrogen sulfate);(1) (1071-29-0)
Carbon disulfide: Carbon disulfide; (75-15-0)
1,3-Thiazinane-2-thione: 2H-1,3-Thiazine-2-thione, tetrahydro-;(2) (5554-48-3)
Triethylamine: Ethanamine, N,N-diethyl-; (121-44-8)
Propionyl chloride: Propanoyl chloride; (79-03-8)
N-Propanoyl-1.3-thiazinane-2-thione: 1-Propanone, 1-(dihydro-2-thioxo-2H-1,3-thiazin-3(4H)-yl)-;(3) (2138126-72-2)

|
Stuart C. D. Kennington, born in 1992 in Cambridgeshire, England, received his MChem degree from the University of Warwick in 2015. He is currently carrying out his Ph.D.thesis under the supervision of Prof. Fèlix Urpí and Pedro Romea at the University of Barcelona with a FI scholarship from the Generalitat de Catalunya. His research focuses on new catalyzed asymmetric synthesis methodologies and their application to the total synthesis of natural products. |

|
Oriol Galeote, born in Barcelona in 1997, received his Degree in Chemistry from the University of Barcelona in 2019. He collaborated as an undergraduate internship in the direct and asymmetric construction of carbon-carbon bonds from N-acyl-1,3-thiazinane-2-thiones catalyzed by nickel(II) complexes under the supervision of Prof. Fèlix Urpí and Pedro Romea. He is currently enrolled in the Master in Organic Chemistry of the University of Barcelona. |

|
Miguel Mellado-Hidalgo, born in Barcelona in 1996, received his Degree in Chemistry from the University of Barcelona in 2018. Then, he enrolled in the Master in Organic Chemistry in the same university, joining the group of Prof. Fèlix Urpí and Pedro Romea to study new direct and enantioselective aldol reactions from N-acyl-1,3-thiazinane-2-thiones catalyzed by nickel(II) chiral complexes. Currently, he is carrying out his PhD Thesis under their supervision, focusing his research on new catalytic and asymmetric methods and their application to the synthesis of natural products. |

|
Pedro Romea completed his B.Sc. in Chemistry at the University of Barcelona and followed Ph.D. studies from 1987 to 1991 under the supervision of Professor Jaume Vilarrasa at the same University of Barcelona. Then, he joined the group of Professor Ian Paterson at the University of Cambridge (UK), where he participated in the total synthesis of oleandolide. Back to the University of Barcelona, he became Associate Professor in 1993. His research interests have focused on the development of newsynthetic methodologies and their application to the stereoselective synthesis of naturally occurring molecular structures. |

|
Felix Urpi received his B.Sc. in Chemistry at the University of Barcelona and completed Ph.D. studies under the guidance of Professor Jaume Vilarrasa at the University of Barcelona in 1988. He then worked as a NATO postdoctoral research associate in titanium enolate chemistry with Professor David A. Evans, at Harvard University in Cambridge, MA. He moved back to the University of Barcelona and he became Associate Professor in 1991, where he holds a chair of Full Professor in Organic Chemistry since 2017. His research interests have focused on the development of new synthetic methodologies and their application to the stereoselective synthesis of naturally occurring molecular structures. |

|
Dr. Zhaobin Han received his B.S. degree in chemistry from Nanjing University in 2003. He received his Ph.D. degree from Shanghai Institute of Organic Chemistry under the supervision of Prof. Kuiling Ding and Prof. Xumu Zhang in 2009, working on development of novel chiral ligands for asymmetric catalysis. At present he is an Associate Professor in the same institute, and his current research interests focus on the development of efficient catalytic methods based on homogeneous catalysis. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved