1. Procedure (Note 1)
C. (((1R,3S,3'S)-3,3'-Diethyl-3H,3'H-1,1'-spirobi[isobenzofuran]-7,7'-diyl)-bis(oxy))bis(diphenylophosphane) (3) . An oven-dried (Note 2) 50-mL single-necked round-bottomed flask (14/20 joint) is equipped with a Teflon coated magnetic stir bar (3.2 x 12.7 mm, round). The flask is sealed with a rubber septum, connected to a Schlenk line via a needle adapter and subsequently cooled to 23 °C (Note 3). (1R,3S,3'S)-3,3'-Diethyl-7,7'-bis (methoxymethoxy)-3H,3'H-1,1'-spirobi[isobenzofuran] (2) (1.88 g, 4.69 mmol, 1.00 equiv) (Figure 7A) and methanol (9.4 mL) (Note 37) are added to the flask under nitrogen, and the solution is sonicated (Note 38) for 1 min (Note 39) (Figure 7B). The solution is cooled to 0 °C (Note 7) while stirring (Note 6), and the acetyl chloride (667 µL, 9.39 mmol, 2.00 equiv) (Note 40) is added with a micro-syringe (Note 41) over 10 min at 0 °C (Note 7) (Figure 7C). The ice bath is removed and the reaction mixture is stirred at 23 °C (Note 3) for 130 min (Note 42) (Figure 7D). Saturated NaHCO3 (Note 33) aqueous solution (10 mL) is added to the solution (Note 43), and the mixture is concentrated to about half of its original volume with the aid of a rotary evaporator (Note 19). The mixture is then transferred to a 60-mL separatory funnel (Note 17) and the flask is rinsed with DCM (5 mL) (Notes 16). The organic layer is collected, and the aqueous layer is extracted with DCM (4 x 10 mL) (Notes 16). The combined organic phases are dried with Na2SO4 (10 g) (Note 44), filtered (Notes 14 and 15) into a 100-mL single-necked pear-shaped flask (14/20 joint) with DCM washings (3 x 5 mL) (Notes 16). The solution is concentrated with the aid of a rotary evaporator (Note 19) to afford crude pale, yellow solids (Note 45).

Figure 7. A) Starting material (2) in the reaction flask; B) reaction solution after sonication; C) reaction solution after AcCl addition; D) reaction solution after completion (photo provided by submitters)
An oven-dried (Note 2) 100-mL single-necked round-bottomed flask (24/40 joint) equipped with a Teflon coated magnetic stir bar (9 x 18 mm, egg-shape) is cooled to 23 °C (Note 3) in a desiccator. The crude solids (1.00 g, 3.20 mmol, 1.00 equiv) and DMAP (58 mg, 0.48 mmol, 15 mol%) (Note 46) are added to the flask (Figure 8A) and then sealed with a rubber septum, connected to a Schlenk line with a needle adapter, and purged to a nitrogen atmosphere. Dichloromethane (32 mL) (Notes 16) and triethylamine (1.78 mL, 12.80 mmol, 4.00 equiv) (Note 47) are added to the flask (Figure 8B), and the solution is cooled to 0 °C (Note 7) while stirring (Note 6). Chlorodiphenylphosphine (1.69 g, 7.68 mmol, 2.40 equiv) (Note 48) is added to the solution over 30 min at 0 °C (Note 7) (Figure 8C) with the syringe pump (Note 11). The reaction mixture is stirred at to 23 °C (Note 3) for 8 h (Note 49) (Figure 8D).

Figure 8. A) Starting material and DMAP in the reaction flask; B) reaction solution in DCM; C) addition of PPh2Cl; D) reaction solution after 8 hours (photo provided by submitters)
The stir bar is removed, and the solution is concentrated with the aid of a rotary evaporator (Note 19). The crude product is purified by chromatography on deactivated silica (Note 50) to afford (((1R,3S,3'S)-3,3'-diethyl-3H,3'H-1,1'-spirobi[isobenzofuran]-7,7'-diyl)bis(oxy))bis(diphenylphosphane) 3 (1.68 g, 68.4%, 99% purity) as a white foam (Notes 51 and 52) (Figure 9).
Figure 9. Product 3 (photo provided by submitters)
2. Notes
1. Prior to performing each reaction, a thorough hazard analysis and risk assessment should be carried out with regard to each chemical substance and experimental operation on the scale planned and in the context of the laboratory where the procedures will be carried out. Guidelines for carrying out risk assessments and for analyzing the hazards associated with chemicals can be found in references such as Chapter 4 of "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at
https://www.nap.edu/catalog/12654/prudent-practices-in-the-laboratory-handling-and-management-of-chemical. See also "Identifying and Evaluating Hazards in Research Laboratories" (American Chemical Society, 2015) which is available via the associated website "Hazard Assessment in Research Laboratories" at
https://www.acs.org/content/acs/en/about/governance/committees/chemicalsafety/hazard-assessment.html. In the case of this procedure, the risk assessment should include (but not necessarily be limited to) an evaluation of the potential hazards associated with
1,3,5-trimethoxybenzene,
3-(methoxymethoxy)-benzaldehyde,
4-(dimethyl-amino)pyridine,
acetic acid,
acetyl chloride,
ammonium chloride,
benzene,
chlorodiphenylphosphine,
chloroform,
dibromomethane,
diethyl carbonate,
diethylzinc,
ethyl acetate,
hexanes,
hydrochloric acid,
magnesium sulfate anhydrous,
methylene chloride,
n-butyllithium, silica,
sodium bicarbonate,
sodium chloride,
sodium sulfate anhydrous,
THF,
toluene,
triethylamine, as well as the proper procedures for working with dry ice and under an inert atmosphere.
Diethylzinc (neat) is highly pyrophoric, hygroscopic, heat sensitive and a highly water reactive chemical. It should be handled using inert atmosphere techniques and special waste treatment procedure should be followed.
2. Unless otherwise reported, all glassware was dried in a 120 °C oven prior to use and then brought down to 23 °C in a desiccator containing Drierite
TM. The checkers used three-necked flasks in lieu of the single-necked, round-bottomed flasks and equipped the flask with a temperature probe with no change in yield, provided the flask is oven dried in a similar manner and kept under nitrogen as described.
3. 23 °C was determined to be between 22 °C and 23 °C.
4.
Diphenyl((S)-1-((S)-1-phenylethyl) aziridin-2-yl)methanol (
A) was prepared following our procedure described in
Org. Synth. 2021,
98, 446-462.
5.
Hexane (mixture of isomers, anhydrous, ≥99%) was purchased from Sigma-Aldrich and used as received.
6. IKA RET basic hot plate stirrer (115V, 620W, 50-60 Hz) and Cole-Parmer IKA C-Mag hot plate stirrer (115V, 1000W, 50-60 Hz) were used. Unless indicated otherwise, 500 rpm was used for stirring.
7. The temperature of 0 °C was reached and maintained by mixing deionized water with ice.
8.
Diethylzinc (≥52 wt. % Zn basis) was purchased from Sigma-Aldrich and used as received.
9. The checkers equipped the flask containing
A with an oven-dried pressure equalizing addition funnel and transferred the
diethylzinc solution by cannula to the addition funnel, which was used for the slow addition of the
diethylzinc solution. Instead of addition via cannula, an oven-dried addition funnel can be used for the slow addition of
diethylzinc. A three-necked round-bottomed flask should be used in this case (
Note 2). Cannula (Chemglass Life Sciences Supplier Diversity Partner Cannula, Stainless Steel, 18 Gauge X 24" Length, Airfree, Schlenk) was purchased from Fisher Scientific.
10.
3-(Methoxymethoxy)benzaldehyde (≥95%) was purchased from Toronto Research Chemicals and used as received (CAS# 13709-05-2).
11. The Fisherbrand™ syringe pump was setup with a built-in syringe size table for Air-Tite™ All-Plastic Norm-Ject™ Syringes.
12. The reaction progress can be monitored by TLC (SiO
2,
hexane/
EtOAc (4/1), starting material: R
f 0.57, product
4: R
f 0.41; UV-C 254 nm) to observe complete consumption of starting material (S refers to starting material, C refers to co-spot of reaction mixture and starting materials, and R refers to the reaction mixture (Figure 10)).
Figure 10. TLC monitoring of Step A (photo provided by submitters)
13.
Hydrochloric acid (ACS reagent, 37%) was purchased from Sigma-Aldrich and diluted to 1N with deionized water.
14. Büchner filter funnels, 24/40 lower vacuum assembly, with coarse frit were purchased from Chemglass Life Sciences. For filtration, a vacuum pump supplied by Heidolph was used to establish reduced pressure.
15. To wash the filter cake effectively, vacuum was turned off between separate washing cycles, washing solvent was added and the resultant mixture was stirred thoroughly with a stainless-steel spatula before removal of the washing solvent by vacuum suction.
16. Dichloromethane (HPLC) was purchased from Fisher Scientific and purified by pressure filtration under nitrogen through activated alumina prior to use.
17. Deionized water was obtained and used directly from the university supply.
18.
Magnesium sulfate anhydrous (Powder/Certified) was purchased from Fisher Scientific and used as received.
19. BUCHI™ Rotavapor™ Scholar with Dry Ice Cold Trap Condenser was connected to Heidolph™ Valve-Regulated Vacuum Pump. Unless specified differently, water bath remained at 30 °C and the vacuum was regulated to 20 mmHg.
20. The crude material was loaded onto a slurry-packed (
hexane) column (ID 72 mm) containing SiO
2 (400 g, 40 - 63 μm, 60 Å silica gel purchased from SiliCycle Inc.), and the flask was then rinsed with DCM (20 mL) (
Note 36) which was loaded afterwards. After loading, solvents were eluted under positive nitrogen pressure and fractions were taken in 50-mL tubes. The solvent system was switched to 5000 mL of 4/1
hexane/
EtOAc (ACS grade purchased from Fisher Scientific which was used as received). Product
1 (R
f 0.41,
hexane/EtOAc 4/1, v/v) eluted, and fractions 31 through 98 were combined (Figure 11), concentrated on a rotary evaporator (30 °C, 780 to 20 mmHg), and dried in vacuo (1-2 mmHg) at ambient temperature for 12 h.
Figure 11. TLC analysis of fractions. (Visualization with UV-C 254 nm) (photo provided by submitters)
21. The product
(1) exhibited the following properties: 99% ee (HPLC (Chiralpak IA column, 97:3
hexanes/isopropanol, 1.0 ml/min), tr = 15.4 min (
R), 17.0 min (
S)); [α]
D23 -27.35 (
c 0.30, CHCl
3); R
f 0.41 (4/1,
hexanes/
EtOAc, v/v); bp 89.5 °C (8.5 mmHg); IR (film): 3360 (br), 2972, 2928, 1602, 1590, 1488, 1454, 1240, 1148, 1076,1014, 992, 924 cm
-1;
1H NMR
pdf (500 MHz, CDCl
3) d: 7.28-7.24 (m, 1H), 7.07-7.03 (m,1H), 6.97 (ddd,
J = 17.0, 7.5, 2.4 Hz, 2H), 5.18 (s, 2H), 4.57 (t,
J = 6.6 Hz, 1H), 3.48 (s, 3H), 1.83-1.72 (m, 2H), 0.93 (t,
J = 7.4 Hz, 3H);
13C NMR
pdf (100 MHz, CDCl
3) d: 157.3, 146.6, 129.9, 119.6, 115.1, 114.0, 94.4, 75.7, 55.9, 31.8, 10.1. HRMS (ESI)
m/z calcd for C
11H
17O
3 [M+H]
+ 197.1171, found 197.1175. Purity was determined by quantitative
1H NMR
pdf spectroscopic analysis using biphenyl as an internal standard to be 97% by weight. The checkers also performed this reaction on half the reported scale, which resulted in a 91% yield of product. The ee was determined by HPLC analysis with a Waters Alliance e2695 Separations Module HPLC system equipped with a CHIRALPAK IA column (length 250 mm, I.D. 4.6 mm). Optical rotations were measured at 23 °C in a solvent of choice on a JASCO P-2000 digital polarimeter at 589 nm (D-line).
22.
Toluene (Certified ACS) was purchased from Fisher Scientific and purified by pressure filtration under nitrogen through activated alumina prior to use.
23.
n-Butyllithium solution (2.5 M in
hexanes) was purchased from Sigma-Aldrich, titrated to be 2.42 M, and used after titration with diphenyl
acetic acid.
24. The SyringeONE syringe pump was purchased from Fisher Scientific and was programmed based on specifications of Air-Tite™ All-Plastic Norm-Ject™ Syringes. The checkers recommend addition of the
n-butyllithium solution in two portions using a 100-mL syringe.
25.
THF (HPLC) was purchased from Fisher Scientific and purified by pressure filtration under nitrogen through activated alumina prior to use.
26. The reaction mixture was changed from an off-white thick mixture to orange solution (Figure 12).
Figure 12. A) Reaction suspension at 3 hours after addition of nBuLi; B) reaction mixture during the addition of THF; C) reaction solution after addition of THF (photo provided by submitters)
27.
Diethyl carbonate (99+%, Alfa Aesar) was purchased from Fisher Scientific and was dried with activated 4A molecular sieves prior to use.
28. Overnight throughout this manuscript refers to 14 h.
29.
Acetic acid (Glacial (Certified ACS), Fisher Chemical) was purchased from Fisher Scientific and used as received.
30. The reaction mixture color changed from black to dark orange after the addition of
acetic acid and turned to yellow after 4 h (Figure 13). The checkers observed an exotherm of 7 °C during the 30 min addition of
acetic acid.
Figure 13. A) Reaction mixture during addition of AcOH; B) reaction mixture after addition of AcOH; C) reaction mixture after 4 h of stirring (photo provided by submitters)
31. The reaction progress can be monitored by TLC (SiO
2,
Hexane/
EtOAc 4/1, starting material
1: R
f 0.39, product
2: R
f 0.53; UV-C 254 nm) (S refers to starting material, C refers to co-spot of reaction mixture and starting materials, and R refers to the reaction mixture (Figure 14)).
Figure 14. TLC monitoring of Step B (photo provided by submitters)
32. Fisherbrand™ reusable glass wide-mouth Erlenmeyer flasks were purchased from Fisher Scientific.
33.
Sodium bicarbonate (Powder/Certified ACS, Fisher Chemical) was purchased from Fisher Scientific and used as received.
34. The first 50 g of
NaHCO3 was added in 1-g portions and the last 50 g were added in 2-g portions.
35. The crude material was loaded onto a slurry-packed (
hexane) column (ID 72 mm) containing SiO
2 (500 g, 40 - 63 μm, 60 Å silica gel purchased from SiliCycle Inc.), and the flask was then rinsed with 9/1
hexane/
EtOAc (40 mL), which was loaded afterwards. After loading, solvents were eluted under positive nitrogen pressure and fractions were taken in 50-mL tubes. The solvent system was switched to 1000 mL of 9/1
hexane/EtOAc (ACS grade purchased from Fisher Scientific which was used as received), then 1800 mL of 8/1
hexane/EtOAc, followed by 800 mL of 7/1
hexane/EtOAc, and finished by 1500 mL of 6/1
hexane/EtOAc. Product
2 (R
f 0.53,
hexane/EtOAc 4/1, v/v) eluted first and fractions 60 through 99 were combined (Figure 15), concentrated on a rotary evaporator (30 °C, 780 to 20 mmHg), and dried in vacuo (1-2 mmHg) at ambient temperature for 12 h. Starting material
1 (R
f 0.39,
hexane/EtOAc 4/1, v/v) eluted later and fractions 111 through 130 were combined, concentrated on a rotary evaporator (30 °C, 780 to 20 mmHg), and dried in vacuo (1-2 mmHg) at ambient temperature for 12 h.
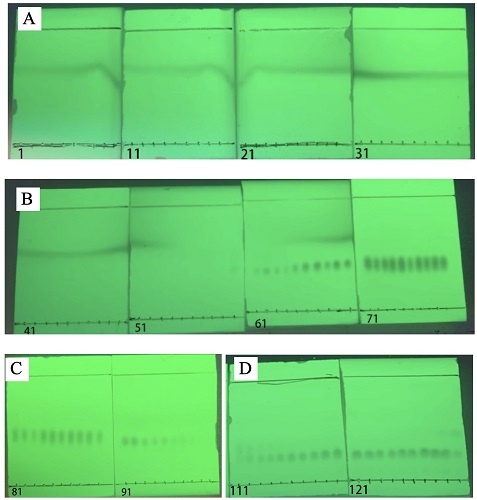
Figure 15. TLC analysis of the fractions. (Visualization with UV-C 254 nm) A) fractions 1 through 40; B) fractions 41 through 80; C) fractions 81 through 100; D) fractions 111 through 130 (photo provided by submitters)
36. The product
(2) exhibited the following properties: 99% ee (HPLC (Chiralpak IA column, 99:1
hexanes/isopropanol, 1.0 ml/min), tr = 12.9 min (
R), 15.2 min (
S)); [α]
D23 +4.76 (
c 2.30, CHCl
3); R
f 0.53 (4/1,
hexanes/
EtOAc, v/v); bp 161.4 °C; IR (film): 2968, 2942, 1616, 1599, 1478, 1258, 1154, 1008, 964, 926, 764 cm
-1;
1H NMR
pdf (500 MHz, CDCl
3) d: 7.28 (t,
J = 7.7 Hz, 2H), 6.87 (dd,
J = 8.3, 4.7 Hz, 4H), 5.40 (dd,
J = 7.3, 3.9 Hz, 2H), 4.94 (d,
J = 6.6 Hz, 2H), 4.82 (d,
J = 6.6 Hz, 2H), 3.07 (s, 6H), 2.03-1.94 (m, 2H), 1.89-1.82 (m, 2H), 1.04 (t,
J = 7.43 Hz, 6H);
13C NMR
pdf (101 MHz, CDCl
3) d: 152.6, 145.7, 130.7, 127.9, 115.9, 114.1, 112.3, 93.4, 83.2, 55.7, 53.5, 28.2, 9.8; Purity was determined by quantitative
1H NMR
pdf spectroscopic analysis using
1,3,5-trimethoxybenzene as an internal standard to be 97% by weight. The checkers also performed this reaction on half the described scale, which resulted in a 67% yield of product. The ee was determined by HPLC analysis with a Waters Alliance e2695 Separations Module HPLC system equipped with a CHIRALPAK IA column (length 250 mm, I.D. 4.6 mm). Optical rotations were measured at 23 °C in a solvent of choice on a JASCO P-2000 digital polarimeter at 589 nm (D-line).
37. Methanol (Certified ACS, Fisher Chemical) was purchased from Fisher Scientific and used as received.
38. Branson® Ultrasonic Bath (115 Vac, 60 Hz) was used with 2.8 L (0.75-gal) tank filled with water at 23 °C.
39. The solution should be homogeneous or more time would be needed in the ultrasonic machine.
40.
Acetyl chloride (reagent grade, 98%) was purchased from Sigma Aldrich and used as received.
41. All micro-syringes (Chemglass Life Sciences, Gas-Tight, Fixed Needle, 22s Gauge) were purchased from Fisher Scientific. A 1 mL syringe may be substituted for a micro syringe if needed.
42. The reaction can be monitored by TLC (SiO
2,
Hexane/
EtOAc 5/1, starting material
2: R
f 0.49, product: R
f 0.32; UV-C 254 nm) (S refers to starting material. C refers to co-spot of reaction mixture and starting materials. R refers to the reaction mixture (Figure 16)).
Figure 16. TLC monitoring of Step C, Part 1 (photo provided by submitters)
43. The resulting solution was shown slight basic (pH = 8) by the pH paper.
44.
Sodium sulfate anhydrous (Granular/Certified ACS) was purchased from Fisher Scientific and used as received.
45. Only 1.00 g of crude product (Figure 17) was used for the remaining step of the ligand synthesis.
Figure 17. Crude material from the deprotection step (photo provided by submitters)
46.
4-(Dimethylamino)pyridine (≥99%) was purchased from Sigma Aldrich and used as received.
47.
Triethylamine (99%) was purchased from Fisher Scientific and distilled under nitrogen from sodium hydride before the use. The use of dry
triethylamine is recommended, KF < 200 ppm.
48.
Chlorodiphenylphosphine (96%) was purchased from Sigma Aldrich, stored in the glovebox and used as received.
49. The reaction can be monitored by TLC (SiO
2,
Hexane/
EtOAc 5/1, starting materials: R
f 0.19, product
3: R
f 0.53; UV-C 254 nm) (S refers to starting materials. C refers to co-spot of reaction mixture and starting materials. R refers to the reaction mixture (Figure 18)).
Figure 18. TLC monitoring of Step C, Part 2 (photo provided by submitters)
50. The crude was loaded onto a slurry-packed (
hexane with 1% TEA) column (ID 42 mm) containing SiO
2 (120 g, 40 - 63 μm, 60 Å silica gel purchased from SiliCycle Inc.), and the flask was then rinsed with
chloroform (3 mL) which was loaded afterwards. After loading, solvents were eluted under positive nitrogen pressure and fractions were taken in 25-mL tubes. The solvent system was switched to 1.1 L of 25/1
hexane (with 1% TEA)/
EtOAc (ACS grade purchased from Fisher Scientific which were used as received), followed by 500 mL of 20/1
hexane (with 1% TEA)/
EtOAc, and then 500 mL of 10/1
hexane (with 1% TEA)/
EtOAc. Product
3 (R
f 0.52,
hexane/
EtOAc 5/1, v/v) eluted, fractions 56 through 96 were combined (Figure 19), concentrated on a rotary evaporator (30 °C, 780 to 20 mmHg), and dried in vacuo (1-2 mmHg) at ambient temperature for 12 h.
Figure 19. TLC analysis of fractions 1 to 96. (Visualization with UV-C 254 nm) (photo provided by submitters)
51. The product
3 exhibited the following properties: [α]
D23 -108.77 (
c 0.20,
toluene); R
f 0.52 (5/1,
hexanes/
EtOAc, v/v); mp 159.4-160.1 °C; IR (film, ATR): 3049, 2967, 2926, 2874, 1594, 1471, 1435, 1344, 1283, 1247, 1091, 1065, 1020, 1006, 994, 937, 831, 771, 730, 701, 652, 633 cm
-1;
1H NMR
pdf (500 MHz,
benzene-d6) d: 7.39-7.35 (m, 4H), 7.25 (dd,
J = 5.2, 2.5 Hz, 2H), 7.16-7.11 (m, 4H), 7.02-6.96 (m, 8H), 6.95-6.92 (m, 6H), 6.57 (d,
J = 7.5, 2H), 5.35 (dd,
J = 8.8, 3.7 Hz, 2H), 1.57-1.43 (m, 4H), 0.93 (t,
J = 7.4 Hz, 6H);
13C NMR
pdf (126 MHz,
benzene-d6) d: 153.0, 152.9, 147.4, 141.1, 141.0, 140.9, 140.8, 131.1, 131.0, 130.8, 130.7, 130.2, 130.0, 129.9, 129.8, 129.2, 128.8, 128.7, 128.7, 128.7, 128.5, 115.7, 115.6, 115.5, 115.0, 83.2, 28.6, 10.8;
31P NMR
pdf (202 MHz,
benzene-d6) d: 105.3. HRMS (ESI)
m/z calcd for C
43H
39O
4P
2+ 681.2317 [M+H]
+ found 681.2316. When the checkers performed the reaction a second time on full scale, 1.49 g (68%) of the product was isolated. Purity was determined by quantitative
1H NMR
pdf spectroscopic analysis using
1,3,5-trimethoxybenzene as an internal standard to be 97% by weight.
52. The compound
3 should be stored in the glove box after purification.
3. Discussion
Figure 20. Selected example of privileged chiral ligands and scaffolds
Scheme 1. Synthesis of SPINOL with racemic intermediate
Scheme 2. Enantioselective synthesis of SPIROL
Scheme 3. Iridium-catalyzed hydroarylations with (S,S,S)-SPIRAP
Scheme 4. Application to enantioselective synthesis of (-)-(R)-angustureine
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved