Org. Synth. 2008, 85, 118
DOI: 10.15227/orgsyn.085.0118
CATALYTIC ENANTIOSELECTIVE ADDITION OF TERMINAL ALKYNES TO ALDEHYDES: PREPARATION OF (S)-(-)-1,3-DIPHENYL-2-PROPYN-1-OL AND (S)-(-)-4-METHYL-1-PHENYL-2-PENTYN-1,4-DIOL
Submitted by Ryo Takita, Shinji Harada, Takashi Ohshima, Shigeki Matsunaga, and Masakatsu Shibasaki
1.
Checked by Shaun Fontaine, Julia Robinson, and Rick L. Danheiser.
1. Procedure
A. (S)-(-)-1,3-Diphenyl-2-propyn-1-ol (3). A flame-dried, 50-mL, two-necked, round-bottomed flask equipped with a glass stopper, argon inlet adapter, and teflon-coated magnetic stir bar is charged with (S)-BINOL (0.230 g, 0.80 mmol) (Note 1) and then is transferred to a glove box and charged with InBr3 (0.291 g, 0.80 mmol) (Note 2). The flask is removed from the glove box and equipped with a rubber septum and a flame-dried reflux condenser fitted with an argon inlet adapter. Dichloromethane (4.0 mL) (Note 3) and then benzaldehyde (4.1 mL, 40 mmol) (Note 4) are added via syringe at room temperature. The resulting solution is stirred at 20–25 °C for 15 min, after which dicyclohexylmethylamine (0.86 mL, 4.0 mmol) (Note 5) is added by syringe. After 10 min, phenylacetylene (8.8 mL, 80 mmol) (Note 6) is added in one portion by syringe and the resulting yellow solution is stirred at 40 °C (oil bath temperature) for 48 h (Note 7). The reaction mixture is then allowed to cool to room temperature, then is poured into a 200-mL separatory funnel (Note 8), and is washed with 50 mL of aqueous 1 M HCl solution. The aqueous phase is separated and extracted with two 50-mL portions of dichloromethane, and the combined organic layers are dried over Na2SO4 (2 g) (Note 9), filtered, and concentrated by rotary evaporation (room temperature, 20 mmHg) to afford 13.1 g of a yellow oil. This material is purified by silica gel column chromatography (elution with ethyl acetate/hexanes) (Note 10) to furnish 6.10–6.45 g (73-77%) of 3 as a pale yellow solid (Note 11). The enantiomeric excess is determined to be 98% by CSP-HPLC (Note 12).
B. (S)-(-)-4-Methyl-1-phenyl-2-pentyn-1,4-diol (5). A flame-dried, 50-mL, two-necked, round-bottomed flask equipped with a glass stopper, argon inlet adapter, and Teflon-coated magnetic stir bar is charged with (S)-BINOL (0.573 g, 2.0 mmol) (Note 1) and then transferred to a glove box and charged with InBr3 (0.713 g, 2.0 mmol) (Note 2). The flask is removed from the glove box and equipped with a rubber septum and a flame-dried reflux condenser fitted with an argon inlet adapter. Dichloromethane (10 mL) (Note 3) and then benzaldehyde (4.1 mL, 40 mmol) (Note 4) are added via syringe at room temperature. The resulting solution is stirred at 20–25 °C for 15 min, after which dicyclohexylmethylamine (2.1 mL, 10 mmol) (Note 5) is added by syringe. After 10 min, 2-methyl-3-butyn-2-ol (4) (7.8 mL, 80 mmol) (Note 13) is added in one portion by syringe and the resulting yellow solution is stirred at 40 °C (oil bath temperature) for 48 h (Note 14). The reaction mixture is then allowed to cool to room temperature, poured into a 200-mL separatory funnel (Note 8), and washed with 50 mL of aqueous 1 M HCl solution. The aqueous phase is separated and extracted with two 50-mL portions of dichloromethane, and the combined organic layers are dried over Na2SO4 (Note 9), filtered, and concentrated to afford 13.6 g of a yellow oil. This material is purified by silica gel column chromatography (elution with ethyl acetate/hexanes) (Note 15) to furnish 6.37–6.87 g (83-89%) of 5 as a colorless solid (Note 16). The enantiomeric excess is determined to be 99% by CSP-HPLC (Notes 17 and 18).
2. Notes
1.
(S)-BINOL (chemical purity 99.8%, enantiomeric purity 99.8%) was purchased by the submitters from Fujimoto Bunshi Kagaku and by the checkers from Aldrich Chemical Company, Inc. (chemical purity 99%, enantiomeric purity 99%). The reagent was further purified by recrystallization from diethyl ether/hexane according to the following procedure.
(S)-BINOL (5.0 g) was dissolved in hot diethyl ether (60 mL) and hexane (100 mL, room temperature) was added. The solution was allowed to stand at room temperature for 2 d and the resulting colorless crystals were collected by filtration to afford
3.9 g of (S)-BINOL which was used after drying at 60 °C (0.3 mmHg) for 24 h. The material purchased by the submitters contained a pale brown impurity which was removed prior to recrystallization by dissolving
10 g of the (S)-BINOL in
100 mL of ether, adding 3 g of activated charcoal, stirring for 1 h, and then filtering the mixture through Celite
® and concentrating by rotary evaporation (room temperature, 20 mmHg).
2.
InBr3 (99.999%) was purchased from Aldrich Chemical Company, Inc. and used without further purification. InBr
3 must be stored and handled in a glove box as the quality of the reagent was found to be important for the success of the reaction.
3.
Anhydrous dichloromethane was purchased by the submitters from Kanto Chemical and used without further purification. The checkers obtained
dichloromethane from Mallinckrodt Baker (>99.8%) and purified it by pressure filtration through activated alumina. In general, the solvent concentration for this reaction is set to 0.2 M based on InBr
3 (5 mL of solvent per 1 mmol of InBr
3).
4.
Benzaldehyde (98+%) was purchased by the submitters from Wako Pure Chemical Industries and distilled from CaH
2 prior to use. The checkers obtained
benzaldehyde (99.5%) from Aldrich Chemical Company, Inc. and distilled it at 57–58 °C (8.2 mmHg) from CaH
2 the same day it was used.
5.
Dicyclohexylmethylamine (>97.0%) was purchased by the submitters from Tokyo Chemical Industry Co., Ltd. and distilled from CaH
2 prior to use. The checkers obtained
dicyclohexylmethylamine (97%) from Aldrich Chemical Company, Inc. and distilled it at 82-85 °C (110 mmHg) from CaH
2 on the same day it was used.
6.
Phenylacetylene (2) (>97%) was purchased by the submitters from Tokyo Chemical Industry Co., Ltd. and by the checkers from Fluka. The
alkyne (50 mL) was passed through a pad of activated alumina (diameter = 2.5 cm, height = 3.3 cm) and then distilled at 73 °C (77 mmHg). The purity of the alkyne was found to have a large effect on its reactivity in this reaction, and the alkyne is best distilled immediately prior to use. The order of addition (aldehyde first, then amine, and finally alkyne) was also found to be important to obtain the best results.
7.
The reaction was monitored by thin layer chromatography (TLC) on Merck or EMD precoated 0.25 mm silica gel 60 F
254 plates, visualization with 254 nm UV light followed by a
p-anisaldehyde stain, elution with hexane/ethyl acetate/4:1 , R
f (
3) = 0.32, R
f (benzaldehyde) = 0.52.
8.
The flask was washed with small portions of dichloromethane (50 mL total) which were added to the separatory funnel.
9.
Anhydrous sodium sulfate was purchased from Nacalai Tesque, Inc. or Mallinckrodt.
10.
Column chromatography was performed by the checkers using 200 g of Sorbent Technologies 230-400 mesh silica gel 60 (6 × 14 cm column) and 100-mL fractions were collected
(1500 mL of hexane/ethyl acetate, 95:5 and then
3500 mL of hexane/ethyl acetate, 92:8). The desired product was obtained in fractions 21-47, which were combined and concentrated by rotary evaporation at room temperature (20 mmHg) and then dried at 4 mmHg overnight to provide a pale yellow solid. The submitters obtained the product as a pale-yellow oil.
11.
The product exhibits the following physicochemical properties: pale yellow solid; mp 40-42 °C; [α]
21D -1.85 (c 1.25, CHCl
3); IR (neat): 3345, 2229, 1598, 1489, 1454, 1030, 756, 690 cm
−1;
1H NMR
pdf (400 MHz, CDCl
3) δ: 2.38 (d,
J = 6.0 Hz, 1 H), 5.71 (d,
J = 6.4 Hz, 1 H), 7.30-7.51 (m, 8 H), 7.62-7.65 (m, 2 H);
13C NMR
pdf (400 MHz, CDCl
3) δ: 65.3, 86.9, 88.8, 122.6, 126.9, 128.5, 128.6, 128.8, 128.9, 131.9, 140.8; HRMS (ESI (+))
m/z 231.0770 [M+Na]
+; Anal. Calcd. for C
15H
12O: C, 86.51; H, 5.81. Found: C, 86.47; H, 5.73.
12.
Enantiomeric excess was determined to be 98% by HPLC.
tR (minor) 11.0 min,
tR (major) 18.3 min (DAICEL CHIRALCEL OD-H,; hexane/2-propanol, 9:1; 1.0 mL/min; 254 nm, 22 °C).
13.
2-Methyl-3-butyn-2-ol (4) (>98%) was purchased by the submitters from Tokyo Chemical Industry Co., Ltd. and by the checkers from City Chemical Company and was distilled from CaSO
4 (DRIERITE
®) at 100 °C (760 mmHg) prior to use.
14.
The reaction was monitored by thin layer chromatography (TLC) on Merck or EMD precoated 0.25 mm silica gel 60 F
254 plates, visualization with 254 nm UV light followed by a
p-anisaldehyde stain, elution with hexane/ethyl acetate, 2:1, R
f (
5) = 0.29, R
f (benzaldehyde) = 0.71.
15.
Column chromatography was performed by the checkers using 100 g of Sorbent Technologies 230-400 mesh silica gel 60 (5.5 × 11 cm column) and 100-mL fractions were collected
(1000 mL of hexane/ethyl acetate, 85:15, then
1000 mL each of 75:25, 65:45, and finally 60:40 hexane/ethyl acetate). The desired product was obtained in fractions 15-39, which were combined and concentrated by rotary evaporation at room temperature (20 mmHg). The residue was then dissolved in
20 mL of dichloromethane and concentrated by rotary evaporation to remove residual ethyl acetate, and the resulting solid was dried at 4 mmHg overnight.
16.
The product exhibits the following physicochemical properties: colorless solid; mp 69-70 °C (lit.
7 66 °C); [α]
20D -15.4 (c 3.31, CHCl
3); IR (neat) 3335, 2982, 1455, 1165, 950, 699 cm
−1;
1H NMR
pdf (400 MHz, CDCl
3) δ: 1.52 (s, 6 H), 3.15 (br s, 1 H), 3.47 (br s, 1 H), 5.44 (s, 1 H), 7.29-7.38 (m, 3 H), 7.49-7.51 (m, 2 H);
13C NMR
pdf (400 MHz, CDCl
3) δ: 31.3, 31.4, 64.4, 65.4, 82.0, 91.5, 126.9, 128.4, 128.7, 140.7 ; HRMS (ESI (+)
m/z 213.0878 [M+Na]
+; Anal. Calcd. for C
12H
14O
2: C, 75.76; H, 7.42. Found: C, 75.71; H, 7.44.
17.
Enantiomeric excess was determined to be 99% by HPLC;
tR (major) 12.9 min,
tR (minor) 16.4 min (DAICEL CHIRALCEL OD-H, eluent: hexane/2-propanol, 9:1; 0.5 mL/min; 254 nm, 25 °C). The checkers did not observe the presence of this minor isomer).
18.
The submitters reported the further purification of the product by recrystallization which was performed as follows: 6.71 g of
5 was dissolved in warm dichloromethane (12 mL at 35 °C) and to this solution was slowly added 15 mL of hexane over 10 min. The solution was left standing overnight and the resulting solid was collected on a Buchner funnel under vacuum, then was washed with 30 mL of hexane, and dried at 4 mmHg (room temperature) overnight to afford 6.47 g of a white crystalline solid. The enantiomeric excess of this material was determined to be >99% ee by HPLC
(Note 17).
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The addition of terminal alkynes to aldehydes, especially in an enantioselective manner, is of great interest because of the versatility of the corresponding propargylic alcohols.
2 Stoichiometric amounts of strong bases such as organolithium, organomagnesium, or dialkylzinc reagents are widely used for this type of reaction with chiral ligands or chiral Lewis acid complexes. However, intrinsic drawbacks, such as the use of excess amounts of metal reagents and a separate step for metal acetylide preparation, make it difficult to achieve an efficient process. Thus, the use of only catalytic amounts of chiral metal salts and the use of terminal alkynes directly as a substrate under a simple operation is desirable. The first example of a catalytic system (Zn(OTf)
2,
N-methylephedrine, and Et
3N) was reported by Carreira and co-workers.
3 Their system provides the corresponding propargylic alcohols in a highly enantioselective manner. Aromatic aldehydes, however, cannot be used in the catalytic system due to the Cannizzaro reaction.
3aThe procedure herein describes the practical preparation of chiral propargylic alcohols under simple and mild conditions catalyzed by an indium(III)/BINOL complex.
4 The catalytic system was developed based on our concept of bifunctional catalysis.
5 The dual activation of soft pro-nuclophiles (alkynes) and hard electrophiles (aldehydes) by indium(III)
6 enabled a wide substrate generality, especially in terms of aliphatic and aromatic aldehydes. In addition, the simple operation in this procedure with the readily available chiral ligand, BINOL, provides an attractive method for the preparation of optically active propargylic alcohols.
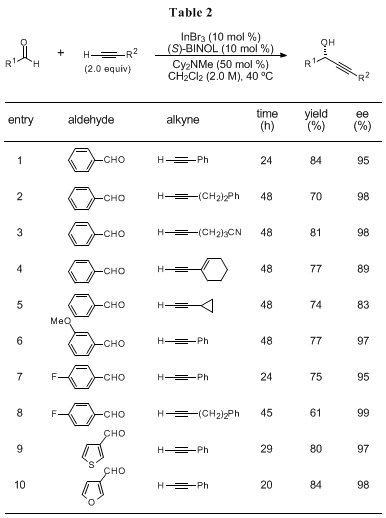
The examples of substrate scope are summarized in Tables 1 and 2.
4a The catalytic system (10 mol %) is applicable for a wide variety of both aliphatic (Table 1) and aromatic aldehydes (Table 2). Phenylacetylene shows high reactivity while the use of alkyl- and alkenylacetylene exhibits slower reaction rates. The limitations of this system should be also noted: the use of acetylene gas, silylacetylenes (e.g. (trimethylsilyl)acetylene), or alkynes having ester functionality (e.g. propargyl propionate) yields no or little amount of the corresponding products under the reaction conditions.
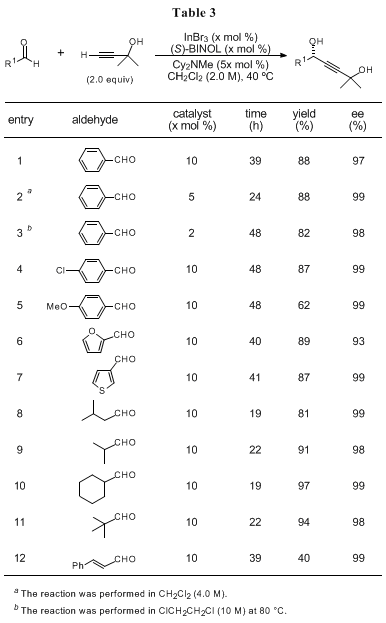
It is noteworthy that the reaction with 2-methyl-3-butyn-2-ol (
4) proceeds smoothly (Procedure B), affording the corresponding products in good chemical yield and high enantioselectivity. In terms of synthetic utility, the use of an acetylene equivalent is fascinating. 2-Methyl-3-butyn-2-ol is the protected form of acetylene, similar to (trimethylsilyl)acetylene, and it is easy to convert the corresponding products to versatile terminal alkynes (i.e., chiral 3-hydroxy-1-alkyne derivatives),
7 which can be further transformed into more complex and useful molecules via Sonogashira coupling reactions, alkylations, and so on. The examples of substrate scope with 2-methyl-3-butyn-2-ol are summarized in Table 3.
4b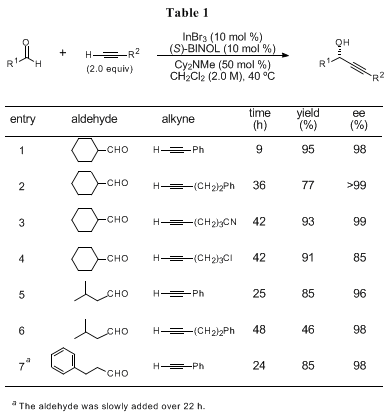
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
(S)-BINOL:
[1,1'-Binaphthalene]-2,2'-diol, (1S)-: (18531-99-2)
Indium bromide: (13465-09-3)
Benzaldehyde: (100-52-7)
(S)-(-)-1,3-Diphenyl-2-propyn-1-ol: Benzenemethanol, α-(2-phenylethynyl)-, (αS)-; (132350-96-0)
Dicyclohexylmethylamine: Cyclohexanamine, N-cyclohexyl-N-methyl-; (7560-83-0)
Phenylacetylene: Ethynylbenzene; (536-74-3)
2-Methyl-3-butyn-2-ol; (115-19-5)
(S)-(-)-4-Methyl-1-phenyl-2-pentyn-1,4-diol: (321855-44-1)
 |
Masakatsu Shibasaki received his PhD. from the University of Tokyo in 1974 under the direction of the late Professor Shun-ichi Yamada before doing postdoctoral studies with Professor E. J. Corey at Harvard University. He joined Teikyo University in 1977, moved to Sagami Chemical Research Center, and took up a professorship at Hokkaido University, before returning to the University of Tokyo as a professor in 1991. He has received many awards, including the Pharmaceutical Society of Japan Award (1999), ACS Award (Arthur C. Cope Senior Scholar Award) (2002), Japan Academy Prize (2005), and ACS Award (Creative Work in Organic Synthesis) (2008). |
 |
Ryo Takita was born in 1978 in Sapporo, Japan. He obtained his PhD from The University of Tokyo in 2006 under the supervision of Professor Masakatsu Shibasaki, working on the development of an indium(III) catalyst system for asymmetric alkynylation of carbonyl compounds. From 2006 to 2007, he worked as a postdoctoral fellow at Massachusetts Institute of Technology with Professor Timothy M. Swager on the synthesis of electroactive hinge molecules. He is currently an assistant professor at Institute for Chemical Research, Kyoto University. |
 |
Shinji Harada was born in 1980 in Tokyo, Japan. After obtaining his B.S. degree from the University of Tokyo in 2002, he received Ph.D. from the University of Tokyo in 2007 under the supervision of Prof. Masakatsu Shibasaki. Then, he joined the faculty at Chiba University, where he is currently an assistant professor in Prof. Atsushi Nishida's group. His research interests are catalytic asymmetric reaction and natural product synthesis. |
 |
Takashi Ohshima received his PhD. from The University of Tokyo in 1996 under the direction of Professor Masakatsu Shibasaki. On the following year, he joined Otsuka Pharmaceutical Co., Ltd. for one year. After two years as a postdoctoral fellow at The Scripps Research Institute with Professor K. C. Nicolaou (1997-1999), he returned to Professor Shibasaki's group as an assistant professor. In 2005, he moved to Osaka University, where he is currently associate professor of chemistry. He has received the Fujisawa Award in Synthetic Organic Chemistry (2001) and Pharmaceutical Society of Japan Award for Young scientists (2004). |
 |
Shigeki Matsunaga was born in 1975 in Kyoto and received his Ph. D. from the University of Tokyo under the direction of Prof. M. Shibasaki. He started his academic career in 2001 as an assistant professor in Prof. Shibasaki's group at the University of Tokyo. He is the recipient of the Yamanouchi Award of Synthetic Organic Chemistry, Japan (2001), and The Chemical Society of Japan Award for Young Chemists (2007). |
 |
Shaun Fontaine (born 1984) graduated with a B.S in Biochemistry and Chemistry from the University of California, San Diego where he carried out undergraduate research in the laboratory of Professor Michael D. Burkart. In 2006 he joined the graduate program at the Massachusetts Institute of Technology and is currently studying the total synthesis of biologically active alkaloids in the laboratory of Professor Rick Danheiser. |
 |
Julia Robinson (born 1984) graduated with a B.A. in Chemistry from Reed College in Portland, Oregon, where she worked in the laboratory of Patrick G. McDougal. She then joined the doctoral program at the Massachusetts Institute of Technology where she is currently an NSF Graduate Fellow pursuing Ph.D. research in the laboratory of Professor Rick Danheiser. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved