Org. Synth. 2012, 89, 19-33
DOI: 10.15227/orgsyn.089.0019
Allylic Oxidation Catalyzed by Dirhodium(II) Tetrakis[ε-caprolactamate] of tert-Butyldimethylsilyl-protected trans-Dehydroandrosterone
Submitted by Maxim O. Ratnikov
1*, Petra L. Goldmann
1, Emily C. McLaughlin
2, and Michael P. Doyle
1.
Checked by Kainan Zhang, Supriyo Majumder, and Kay Brummond.
1. Procedure
Caution! Reactions and subsequent operations involving peracids and peroxy compounds should be run behind a safety shield. Peroxy compounds should be added to the organic material, never the reverse. For relatively fast reactions, the rate of addition of the peroxy compound should be slow enough so that it reacts rapidly and no significant unreacted excess is allowed to build up. The reaction mixture should be stirred efficiently while the peroxy compound is being added, and cooling should generally be provided since many reactions of peroxy compounds are exothermic. New or unfamiliar reactions, particularly those run at elevated temperatures, should be run first on a small scale. Reaction products should never be recovered from the final reaction mixture by distillation until all residual active oxygen compounds (including unreacted peroxy compounds) have been destroyed. Decomposition of active oxygen compounds may be accomplished by the procedure described in Korach, M.; Nielsen, D. R.; Rideout, W. H. Org. Synth. 1962, 42, 50 (Org. Synth. 1973, Coll. Vol. 5, 414). [Note added April 2018].
A. Dirhodium(II) tetrakis[ε-caprolactamate] acetonitrile solvate, Rh2(cap)4(CH3CN)2 (1). An oven-dried 250-mL, single-necked (24/40 joint) round-bottomed flask containing a Teflon-coated stir-bar is charged with dirhodium(II) tetraacetate (2.00 g, 4.5 mmol) (Note 1), e-caprolactam (11.84 g, 104.6 mmol) (Note 2), and dry chlorobenzene (110 mL) (Note 3). The flask is fitted with a Soxhlet extraction apparatus (Note 4) containing a thimble with 30 g of an oven-dried Na2CO3 and sand mixture in a 3:1 ratio (Note 5). An additional 50 mL of chlorobenzene is added to partially fill the thimble-chamber of the Soxhlet apparatus. The mixture is refluxed at 185 °C (external temperature) open to the atmosphere (Note 6), and after only 15 min the color of the solution turns from green to dark blue. After 10 h the vessel is removed from the oil bath, the Soxhlet extraction apparatus is removed, and the solvent is evaporated under reduced pressure using a rotary evaporator to yield a blue, glass-like solid residue. 2-Propanol (100 mL) (Note 7) is added to the same flask, and the solid residue is mixed vigorously with 2-propanol for 30 seconds using a metal spatula to dissolve chlorobenzene that is entrapped in the residue (Note 8). The 2-propanol is evaporated from the solid residue under reduced pressure to give a purple powder suitable for filtration (Note 9). Acetone (100 mL) is added to dissolve excess e-caprolactam contained in the powder, and the solid is gravity-filtered through a Büchner funnel and then washed with an additional 50 mL of acetone (Note 10). The filtered purple solid is transferred to a pre-weighed, 250-mL round-bottomed flask and dried under high vacuum (0.1 mmHg) for 1 h to yield 3.52 g of a dull purple powder (Note 11). To render the powder catalytically active (Note 12) the solid is recrystallized from 66 mL of boiling acetonitrile/methanol (10:1) (Note 13) to give 1.65 g (50% yield) of Rh2(cap)4(CH3CN)2 (1) (Note 14) as shiny deep purple crystals.
B. Androst-5-en-17-one, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3β)- preparation (3β). A 250-mL, single-necked (24/40 joint) Erlenmeyer flask is dried at about 200 °C under vacuum (0.01 mmHg) for approximately 10 min. The flask is charged under the atmosphere of dry nitrogen with 2 (5.00 g, 17.3 mmol) (Note 15) and 1H-imidazole (1.77 g, 26.0 mmol) (Note 16), followed by N,N-dimethylformamide (25.0 mL) (Note 17). The resulting suspension is sonicated at 20 °C with gentle swirling until homogeneous (Note 18). tert-Butyldimethylsilyl chloride (3.39 g, 22.5 mmol) (Note 19) is added as a solid to the reaction mixture. The flask is equipped with a magnetic stir bar (Note 20). After about 3 min of stirring, a white precipitate begins to form. The reaction is stirred for 30 min, at which time water (100 mL) is added to ensure complete product precipitation. The white clay-like substance is scraped from the glass walls of the Erlenmeyer flask, filtered under vacuum, and then dried overnight under vacuum (0.01 mmHg) at 20 °C to yield 6.83 g (98%) of 3 as a white powder (Note 21).
C. (3β)-3-[[(1,1-dimethylethyl)dimethylsilyl]-oxy]-Androst-5-ene-7,17-dione (4). A 200-mL, Erlenmeyer flask (24/40 joint) containing a Teflon-coated magnetic stir bar is charged with 3 (8.04 g, 20.0 mmol), 1 (146 mg, 0.20 mmol, 1.00 mol %), and 1,2-dichloroethane (40 mL) (Note 22). The suspension is sonicated for 5 min at 20 °C with occasional gentle swirling (Note 18). After addition of T-HYDRO (14.3 mL, 99.5 mmol, 5.0 equiv) in one portion (Note 23), the flask containing the reaction mixture is equipped with a septum containing an outlet needle (Note 24) and its contents are stirred at 40 °C (external temperature) (Note 25). The color of the solution turns from purple to dark red after about 2 min following addition of T-HYDRO. After 24 h the mixture is concentrated on a rotary evaporator (30 °C, 10 mmHg) (Note 26). The product is purified by column chromatography (Note 27). Fractions containing the product are combined, and the solvent is evaporated under reduced pressure on a rotary evaporator (30 °C, 30 mmHg). The remaining white solid is dissolved in diethyl ether (20 mL) at room temperature. The solution is heated to 35 °C and then cooled to room temperature whereupon a white powder crystallizes from the solution. Diethyl ether is removed using a pipette. The above washing procedure is repeated twice. The resulting white powder is collected and dried overnight under reduced pressure at room temperature (60 °C, 4 mmHg) to yield 4 (5.50 g, 66 %) (Notes 28 and 29).
2. Notes
1.
Dirhodium(II) tetraacetate was obtained from Pressure Chemicals Co. and used as received.
2.
e-Caprolactam, 99%, was obtained from Alfa Aesar and used as received.
3.
Chlorobenzene, 99+%, was obtained from Acros and stored over activated 3Å molecular sieves prior to use.
4.
A Soxhlet apparatus (PYREX
®, 31 mm internal diameter × 135 mm external length), which contained a cellulose single thickness extraction thimble obtained from Whatman
® (33 mm internal diameter × 94 mm external length), was used.
5.
Sodium carbonate was used to trap acetic acid liberated in the ligand exchange reaction as sodium acetate/sodium bicarbonate. A piece of glass wool was placed at the top and the bottom of the carbonate-sand mixture to prevent its entry into the reaction flask.
6.
The temperature of the oil bath was maintained at 185-195 °C (external temperature). The Soxhlet extractor was covered with aluminum foil for insulation to provide a higher rate for solvent turnover. Solvent turnover in the Soxhlet extractor (about 60 mL each time) occurred every 30-40 minutes.
7.
Commercial ACS grade 2-propanol was used as received.
8.
Complex
1 has low solubility in 2-propanol.
9.
If the solid remains a slurry, two additional 2-propanol washes of 100 mL each are performed.
10.
Vacuum filtration is applied when washing with commercial ACS-grade acetone.
11.
The dull purple powder was identified as Rh
2(cap)
6 with two axially bound ε-caprolactam ligands:
Calcd. for C
36H
62N
6O
6Rh
2: C, 49.09; H, 7.10; N, 9.54. Found: C, 49.55; H, 6.82; N, 9.29. The analysis was not confirmed by the checkers.
12.
The two axially-ligated ε-caprolactam molecules are tightly bound; they inhibit formation of the catalytically active dirhodium complex.
13.
Recrystallization procedure: In a 100-mL, single-necked (24/40 joint) round-bottomed flask, equipped with a refluxing condenser and a Teflon-coated stir-bar, crude Rh
2(cap)
4 (701.1 mg) is stirred in acetonitrile (40 mL) and methanol (4 mL). The flask is placed in an oil bath maintained at 100 °C (external temperature). When the mixture reaches boiling temperature, additional methanol is added dropwise via Pasteur pipette from the top of the refluxing condenser until a deep purple homogenous solution is obtained. The flask is removed from the oil bath, and the hot solution is vacuum filtered through a funnel containing a small cotton plug. (The submitters caution against the use of vacuum filtration. The recommend gravity filtration with minimal cooling until filtration is complete.) The solution is allowed to cool to room temperature and then placed into a freezer overnight. A shiny purple crystalline solid is isolated by removal of the supernatant liquid (using a pipette). The solid is washed with acetonitrile (3 x 5 mL) and dried under vacuum at 4 mmHg for 8 h.
14.
Data for Rh
2(cap)
4(CH
3CN)
2:
1H NMR (400 MHz, CDCl
3) δ: 1.39-1.67 (m, 26 H), 2.07 (s, 11 H), 2.31-2.43 (m, 8 H), 3.34-3.23 (m, 8 H), The
1H NMR
pdf spectral regions 1.39-1.67 and 2.07 do not integrate to the correct number of protons; however, the data, including relative integration values, are in agreement with those provided by the submitter;
13C NMR
pdf (100 MHz, CDCl
3) δ: 2.3, 24.3, 29.0, 30.7, 36.8, 53.3, 115.9, 185.7; IR (film) 3383, 2958, 2918, 2847, 2359, 2339, 1683, 1596, 1434, 1348 cm
-1; HRMS (ESI) Calcd. for C
24H
40N
4O
4Rh
2: 654.1160, found 654.1180 [M
+- 2(CH
3CN]); Anal. Calcd. for C
28H
46N
6O
4Rh
2: C, 45.66; H, 6.30; N, 11.41; Found: C, 45.65; H, 6.44; N, 11.90. The checkers were unable to obtain satisfactory CHN analysis (checker's results: Anal. Calcd. for C
28H
46N
6O
4Rh
2: C, 45.66; H, 6.30; N, 11.41; Found: C, 42.31; H, 6.48; N, 8.23); however, the oxidation in Step C proceeded efficiently and without difficulty using the catalyst prepared by the checkers.
15.
trans-Dehydroandrosterone (99%) was obtained from Sigma-Aldrich. and used as received.
16.
1
H-Imidazole (ACS reagent, =99%) was obtained from Sigma-Aldrich and used as received.
17.
N,N-Dimethylformamide (99.8 %, anhydrous) was obtained from Aldrich, stored under nitrogen, and used as received.
18.
Sonication facilitates dissolution of starting materials. Steroids
2 and
3 form solid clumps upon addition that have a low dissolution rate when stirring with a magnetic stir bar.
19.
tert-Butyldimethylsilyl chloride (reagent grade, 97 %) was obtained from Aldrich and used as received.
20.
Use of a mechanical stirrer is necessitated by the formation of a large amount precipitate that significantly increases the viscosity of the reaction mixture.
21.
The product has following properties. mp 148-149 °C, lit.
3 value 146-147 °C; [α]
D18 +3.4° (
c 1.0, CHCl
3); lit.
3 [a]
D23 = +7.7° (
c 1.0, CHCl
3); IR (CH
2Cl
2): 2949, 2925, 2888, 2855, 1744, 1372, 1258, 1094, 841 cm
-1; R
f = 0.58 (EtOAc/hexane 1:4);
1H NMR
pdf (400 MHz, CDCl
3) δ: 0.05(s, 6 H), 0.88 (s, 9 H + 3 H), 0.95-1.09 (m, 5 H), 1.24-1.30 (m, 2 H), 1.41-1.74 (m, 7 H), 1.79-1.97 (m, 3 H), 2.03-2.27 (m, 4 H), 2.45 (dd,
J = 19.2, 8.4 Hz, 1 H), 3.47 (tt,
J = 10.8, 4.8 Hz, 1 H), 5.34 (d,
J = 5.2 Hz, 1 H);
13C NMR
pdf (100 MHz, CDCl
3) δ: -4.6, 13.5, 18.2, 19.4, 20.3, 21.8, 25.9, 30.8, 31.4, 31.5, 32.0, 35.8, 36.7, 37.3, 42.7, 47.5, 50.3, 51.7, 72.4, 120.4, 141.7, 221.0; HRMS (ESI): Calcd. for C
25H
42O
2SiNa (M+Na): 425.2852. Found: 425.2846; Anal. Calcd. for C
25H
42O
2Si: C, 74.56; H, 10.51. Found: C, 74.78; H, 10.70.
22.
1,2-Dichloroethane (spectrophotometric grade, =99%) was obtained from Aldrich and used as received.
23.
T-HYDRO, a 70% solution of
tert-butyl hydroperoxide in water, was obtained from Aldrich and used as received. A smaller excess of T-HYDRO leads to an incomplete conversion.
24.
The needle provides a gas outlet that prevents a pressure built-up in the reaction mixture. The septum limits solvent evaporation.
25.
The reaction can be monitored by silica gel TLC (Whatman, 200 µm, with F-254) with EtOAc-hexane (1:4) as an eluent and visualized with KMnO
4. Compound
4 has an R
f of 0.49 in EtOAc-hexane (1:4).
26.
If the residue is a syrup, the residue should be diluted with a dichloromethane prior to transfer onto the chromatography column.
27.
The column (5 cm diameter × 20 cm height) of silica gel (95 g of silica gel; Sorbent Technologies, 40-63 µm) is packed as slurry in EtOAc-hexane (1:60) and a layer of sand (1 cm) is added on top. The concentrated reaction mixture is placed on the top of the column and is eluted with EtOAc-hexane mixture of the following ratios and volumes:
|
|
EtOAc-hexane Ratio
|
Volume, mL
|
|
|
1:60
|
160
|
|
1:30
|
40
|
|
1:2
|
400
|
|
The product is collected in fractions 15 to 28 in 13*100 Fisher test tubes. The presence of
4 can be monitored by TLC on silica and visualized with UV light (254 nm) or KMnO
4 (see note 25).
28.
The product has following properties: mp 124-125 °C, lit.
4 value 127-129 °C; [a]
D17 = - 53.5° (
c 1.0, CHCl
3), lit.
4 [a]
D23 = - 51.6° (
c 1.86, CHCl
3); IR (solid): 2949, 2880, 2857, 1740, 1671, 1629, 1462, 1407, 1379, 1295, 1254, 1190, 1096, 1008, 957, 869, 837, 806, 777 cm
-1; R
f = 0.49 (EtOAc/hexane 1:4);
1H NMR
pdf (400 MHz, CDCl
3) δ: 0.05 (s, 6 H), 0.88 (s, 13 H), 1.20 (m, 6 H), 1.48-1.87 (m, 9 H), 1.92 (dt,
J = 13.5, 3.4 Hz, 1 H), 2.07-2.17 (m, 1 H), 2.35-2.50 (m, 4 H), 2.77-2.85 (m, 1 H) 3.60 (tt,
J = 10.4, 5.2 Hz, 1 H), 5.71 (s, 1 H);
13C NMR
pdf (75 MHz, CDCl
3) δ: -4.4, -4.3, 14.1, 17.8, 18.5, 20.9, 24.5, 26.1, 31.1, 32.0, 36.0, 36.7, 38.8, 43.0, 44.7, 46.1, 48.2, 50.5, 71.5, 126.0, 167.2, 201.5, 220.7; HRMS (ESI): Calcd. for (M+H): 417.2825. Found: 417.2798; Anal. Calcd. For C
25H
40O
3Si: C, 72.06; H, 9.68. Found: C, 71.83; H, 9.73.
29.
To address the differences in specific rotation between that reported in footnote 28 and literature values, the
tert-butyldimethylsilyl group is removed with 1.0 M tetrabutylammonium fluoride solution in THF to prepare corresponding hydroxyl derivative and compare its specific rotation with that reported in the literature:
[a]
D21 = - 82.4° (
c 1.0, DCE), lit.
5 [a]
D23 = - 82.8° (methanol).
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
The selective oxidation of hydrocarbons is a topic of increasing importance in organic chemistry and desirability for organic synthesis.
6-22 Allylic oxidation is particularly useful because it generates an α,β-unsaturated carbonyl moiety that is broadly functional in organic syntheses.
23-25 Steroids are prevalent substrates for oxyfunctionalization because the hydrocarbon steroidal core is easily accessible from natural materials, and selective functionalization can be easily achieved.
An oxidative system of great potential has been recently reported: rhodium(II) caprolactamate-catalyzed oxidations by
tert-butyl hydroperoxide (TBHP). The detailed procedure for allylic oxidation and a synthesis of the catalyst are presented in this article. This methodology has proven to be superior in allylic oxidation
26-28 as well as in benzylic
29 and propargylic
30 oxidations, oxidative Mannich reactions of tertiary anilines
31 and oxidations of secondary amines to imines.
32 Some examples of allylic oxidation are presented in Table 1. These reactions give high product yields even when electron poor alkenes (entries 1-3, Table 1) are employed, and this methodology is particularly advantageous in oxidations of steroidal substrates (entries 4-5, Table 1). Although allylic oxidation of 1-substituted-cycloalkenes occurs exclusively at the 3-position in most cases,
24-26 an exception is that of limonene oxidation (Figure 1).
25 Although allylic oxidation of acyclic α,β-unsaturated carbonyl compounds and cyclic alkenes occur in good yield, allylic oxidations of simple acyclic alkenes have not been reported. The mechanism of the reaction is reported to involve formation of allylic radical intermediates that undergo trapping by molecular oxygen.
5Table 1. Examples of allylic oxidation with Rh2(cap)4 / T-HYDRO system.
Figure 1.27 Limonene oxidation
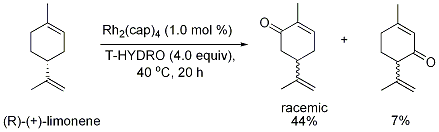
Several methodologies have been developed to achieve efficient allylic oxidation as alternatives to stoichiometric oxidants selenium dioxide
33,34 and chromium(VI).
35-41 Selenium(IV) oxide has the opposite regioselectivity for carbonyl formation of chromium(VI) or of
tert-butyl hydroperoxide oxidations, and both selenium dioxide and chromium(VI) have well-known disadvantages in their applications.
42 As an alternative, procedures have been introduced that employ the mild oxidant
tert-butyl hydroperoxide (TBHP), catalyzed by CrO
3,
43 (
n-Bu
3SnO)
2CrO
2,
44 Cr(CO)
6,
45,46 benzotriazole × CrO
3,
47, ZrOH
2+ × Cr
2O
72-,
48 2PyH
+ × Cr
2O
72-,49,50 C
5H
8N
2H[CrO
3F],
36 RuCl
3,
51 Co(OAc)
2,
52-54 CuI,
55,56 BiCl
3,
57 Mn
3O(OAc)
9,
4 NaClO
2,
58 NaOCl,
59 and Pd(OH)
2.
58,59 Most often excess
tert-butyl hydroperoxide is required because of its catalytic conversion to di-
tert-butyl peroxide, water, molecular oxygen.
25 Typical conditions and reaction outcomes are presented for comparison in Table 2 for the oxidation of cholesteryl acetate by those reactants for which this process has been reported. Chromium reagents and chromium-based catalysts (entries 9-11, Table 2) have limited applicability especially on an industrial scale due to a well-established high toxicity and carcinogenicity of chromium(VI)
42 Reactions catalyzed by chlorine-containing salts (entries 1-2, Table 2) have limited attractiveness for large scale processes due to moderate yields obtained in these oxidation reactions. Some copper- and manganese-based systems (entries 3, 5, Table 2) require large loadings of heavy metals that complicate waste treatment procedures. Several oxidation systems are based on the use of a TBHP solution in decane as an oxidant (entries 3-4, 6-7, Table 2) that is significantly more expensive than the 70% aqueous solution of TBHP (T-HYDRO). Hydrogen peroxide has been reported to effect allylic oxidation promoted by an iron(III) complex (entries 12-13, Table 2), but yields are low and further refinement will be required in order for this oxidant to be effectively employed.
60,61 Use of over 10 equiv of
tert-butyl hydroperoxide per mole of substrate (entries 4, 5, and 8, Table 2) is to be avoided since the increased explosive potential of hydroperoxides must be taken into consideration during scale-up. The rhodium-based process occurs in a high yield with low catalyst and peroxide loadings under mild conditions. The oxidant, T-HYDRO, is inexpensive, and the dirhodium-catalyzed oxidation process is relatively free of competing reactions that include both oxidation of alcohols and alcohol formation.
Table 2. Cholesteryl acetate oxidation
|
|
Entry
|
Catalyst/Additive (mol %)
|
Oxidant
|
Equivalents of Oxidant
|
Time (h)
|
Temp (°C)
|
Yield (%)
|
|
|
1
|
NaClO2 (120%)
|
T-HYDRO
|
5
|
60
|
80
|
6658
|
|
2
|
NaOCl (200%)
|
T-HYDRO
|
6
|
10
|
2 - 5
|
6859
|
|
3
|
Mn3O(OAc)3 (10%)
|
TBHP in decane
|
5
|
48
|
20
|
854
|
|
4
|
BiCl3 (10%)
|
TBHP in decane
|
11
|
22
|
70
|
8257
|
|
5
|
CuI (68%) / n-Bu4N+Br- (12%)
|
T-HYDRO
|
30
|
24
|
40
|
7655
|
|
6
|
CuI (2.6%)
|
TBHP in decane
|
7
|
24
|
70
|
8056
|
|
7
|
Co(OAc)2 immob. on silica (2.2%)
|
TBHP in decane
|
7
|
48
|
70
|
7053
|
|
8
|
RuCl3 × H2O (0.7%)
|
T-HYDRO
|
10
|
24
|
20
|
7551
|
|
9
|
Cr2O72- (immob. On SiO2 / ZrO2)
|
T-HYDRO
|
2
|
n/a
|
20
|
4848
|
|
10
|
Cr2O72- × 2PyH+ (2.5%)
|
T-HYDRO
|
7
|
50
|
40
|
7850
|
|
11
|
Cr(CO)6 (50%)
|
TBHP (90% in water)
|
3
|
15
|
80
|
8045
|
|
12
|
Fe(o-C5H4CO2)3 (50%)
|
H2O2 (30% aq.)
|
3
|
4.5
|
20
|
2060
|
|
13
|
Fe(PA)3 (50%)
|
H2O2 (30% aq.)
|
5
|
3
|
20
|
3461
|
|
14
|
Rh2(cap)4 (1.0%)
|
T-HYDRO
|
5
|
20
|
40
|
8028
|
|
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
ε-Caprolactam: 2H-Azepin-2-one, hexahydro-; (105-60-2)
Dirhodium(II) tetrakis[ε-caprolactamate]: Rhodium, tetrakis[μ-(hexahydro-2H-azepin-2-onato-κN1:κO2)]di-, (Rh-Rh); (138984-26-6)
trans-Dehydroandrosterone: Androst-5-en-17-one, 3-hydroxy-, (3β)-; (53-43-0)
tert-Butyl hydroperoxide: Hydroperoxide, 1,1-dimethylethyl; (75-91-2)
1,2-Dichloroethane: Ethane, 1,2-dichloro-; (107-06-2)
Androst-5-ene-7,17-dione, 3-hydroxy-, (3β)-; (566-19-8)
TBDMSCl: Silane, chloro(1,1-dimethylethyl)dimethyl-; (18162-48-6)
1H-Imidazole; (288-32-4)
N,N-Dimethylformamide: Formamide, N,N-dimethyl-; (68-12-2)
Androst-5-en-17-one, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3β)-; (42151-23-5)
Androst-5-ene-7,17-dione, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3β)-; (157302-54-0)
 |
Michael P. Doyle joined the faculty at Hope College in 1968 and moved to Trinity University in San Antonio, TX, in 1984. In 1997 he came to Tucson, AZ, as Vice President, then President, of Research Corporation and Professor of Chemistry at the University of Arizona. In 2003 he moved to the University of Maryland, College Park, as Professor and Chair of the Department of Chemistry and Biochemistry. His research interests are in catalysis, especially reactions catalyzed by paddlewheeled dirhodium compounds, and in the diverse chemistries that surround the dirhodium framework.
|
 |
Maxim O. Ratnikov was born in Moscow, Russia, in 1984. In 2007 he received his M.S. degree from the Higher Chemical College in Moscow participating in a development of polyfluorinated alcohols as a new electrophilic media under Dr. William Smit's supervision. After receiving his degree, he moved to University of Kentucky where he worked on investigation of bio-inspired nanoparticles under Dr. Marc Knecht's supervision. In 2008, he joined Professor Michael Doyle in University of Maryland where he is currently working towards his doctoral degree. His research interests are in the development of novel oxidative methods and their applications.
|
 |
Petra L. Goldmann was born in 1986 in Munich where she studied chemistry and biochemistry in Ludwig Maximilian University, Munich, Germany and received her B.Sc. degree in 2009. In spring 2009, she did her internship studies with Professor Michael Doyle in University of Maryland, College Park, USA in the development of allylic oxidation catalyzed by dirhodium (II) caprolactamate. Working towards her PhD studies, she is currently participating in elaboration of new organometallic-based synthetic methodology Professor Paul Knochel in Ludwig Maximilian University
|
 |
Emily McLaughlin was born in Pennsylvania and received her B.S. degree from Ohio Northern University in 2001. She completed her Ph.D. studies in total synthesis under Professor Jeffrey Winkler at the University of Pennsylvania in 2006. Soon thereafter, she commenced her post-doctoral training at the University of Maryland with Professor Michael Doyle. In the Doyle lab, she worked on various oxidations catalyzed by dirhodium(II) caprolactamate. Emily is currently an Assistant Professor of Chemistry at Bard College, NY where she and her students are pursuing the design of an aziridination protocol to be applied toward the synthesis of amino acids and heterocyclic scaffolds.
|
 |
Kainan Zhang received his Bachelor's degree in chemical engineering from Hebei University of Science and Technology in 2004. He then joined the graduate program at the Beijing Institute of Technology and obtained a master's degree where he is working on the synthesis of β-lactamase inhibitor Tazobactam. He then moved to the United States and obtained a Ph.D. in Chemistry. During his Ph.D. studies, he worked on transition metal-catalyzed cycloaddition reactions under the guidance of Dr. Janis Louie. He is now a postdoctoral associate and working under the direction of Professor Kay M. Brummond. His current research interests include Rh(I)-catalyzed cyclocarbonylation reactions of allene-ynes.
|
 |
Supriyo Majumder was born in Kolkata, India in 1980. He received his B.Sc. in 2002 from University of Calcutta and M.Sc in chemistry in 2004 from IIT-Madras, India. In 2009 he earned his PhD in chemistry from Michigan State University under the supervision of Professor. Aaron L. Odom. Then he joined the research group of Professor. Kay M. Brummond at University of Pittsburgh and worked in the diversity oriented synthesis using an allene-containing Tryptophan scaffold. Currently, he is working as a research fellow with Professor. Edwin Vedejs at University of Michigan. His current research interests include ionic-hydrogenation with phosphine boranes and intramolecular hydroboration of olefins.
|
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved