Org. Synth. 2013, 90, 10-24
DOI: 10.15227/orgsyn.090.0010
Reagent for Divalent Sulfur Protection: Preparation of 4-Methylbenzenesulfonothioic Acid, S-[[[(1,1-Dimethylethyl)-Dimethylsilyl]oxy]methyl] Ester
Submitted by Lihong Wang and Derrick L. J. Clive
1.
Checked by David Hughes
1. Procedure
Caution: Steps A and B must be conducted in an efficient fumehood.
A. (Ethylthio)methanol (1). An oven-dried 250-mL, 3-necked, round-bottomed flask equipped with a PTFE-coated magnetic stirring bar (3 x 1 cm) is fitted with a gas inlet adapter connected to a nitrogen line and a gas bubbler. The other two necks are capped with rubber septa; a thermocouple probe is inserted through one of the septa (Note 1). Paraformaldehyde (5.58 g, 0.186 mol, 1.07 equiv) (Notes 2 and 3) and ethanethiol (12.8 mL, 10.7 g, 0.173 mol, 1.0 equiv) (Note 4) are added to the flask. The mixture is stirred gently (avoid splashing) and cooled in an ice-water bath to 4 °C. A 25% solution of NaOMe in MeOH (0.07 mL, 0.002 equiv) is added through one of the septa via a 100 μL syringe over 1 min. The temperature rises to 33 °C over 3 min, then cools to 13 °C over 5 min. The solids completely dissolve (Note 5). The ice-water bath is removed and the solution is stirred at 13-16 °C for 15 min. The pale yellow (ethylthio)methanol (1) (Note 6) is used immediately in the next step (Note 7).
B. (1,1-Dimethylethyl)[(ethylthio)methoxy]dimethylsilane (2). Dichloromethane (Note 8) (120 mL) is added to the same flask containing neat (ethylthio)methanol from Step A. The stirred solution is cooled in an ice-water bath to 3 °C, then 4-(dimethylamino)pyridine (0.90 g, 7.4 mmol, 0.04 equiv) and triethylamine (21.9 g, 0.22 mol, 1.27 equiv) are added, followed by chloro(1,1-dimethylethyl)dimethylsilane (30.0 g, 0.20 mol, 1.18 equiv), added in 3 portions over 10 min (Note 9). The ice-water bath is replaced with a water bath and the reaction is stirred for 4 h (Note 10). The mixture is transferred to a 1-L separatory funnel using dichloromethane (60 mL) to rinse the flask. The organic layer is washed with water (2 x 100 mL) and saturated aqueous ammonium chloride (100 mL). The organic phase is filtered through a bed of sodium sulfate (50 g) in a medium porosity sintered glass funnel into a 1-L round-bottomed flask, using dichloromethane (2 x 50 mL) to rinse the filter cake. The solution is concentrated by rotary evaporation (70 mmHg, bath temperature 40 °C). The residue is diluted with hexanes (150 mL), transferred to a 1-L separatory funnel, and washed with water (2 x 200 mL) and brine (75 mL). The organic layer is filtered through a bed of sodium sulfate (50 g) in a medium porosity sintered glass funnel into a 500-mL round-bottomed flask, using hexanes (2 x 50 mL) to rinse the filter cake. The solution is concentrated by rotary evaporation (70 mmHg, bath temperature 40 °C) to afford crude 2 (38 g) as an oil. Vacuum distillation (Note 11) provides (1,1-dimethylethyl)[(ethylthio)methoxy]dimethylsilane (2) as a colorless liquid (28.0-28.6 g, 78-80 % yield over Steps A and B) (Notes 12 and 13).
WARNING: Chloromethyl ethers are potent carcinogens. The preparation and handling of compound 3 should be conducted at all times in a hood or ventilated balance enclosure.
C. (Chloromethoxy)(1,1-dimethylethyl)dimethylsilane (3). An oven-dried 500-mL round-bottomed flask equipped with a PTFE-coated magnetic stirring bar (3 x 1 cm) is capped with a rubber septum pierced with a nitrogen inlet needle connected to a gas bubbler. A thermocouple thermometer probe is also inserted through the septum (Note 1). (1,1-Dimethylethyl)[(ethylthio)methoxy]dimethylsilane (2) (12.1 g, 58.6 mmol, 1.00 equiv) and dichloromethane (120 mL) are added to the flask. The stirred solution is cooled to 2 °C using an ice-water bath. Sulfuryl chloride (8.14 g, 60.3 mmol, 1.03 equiv) (Note 14) is added via a 10 mL syringe over 10 min, keeping the temperature <5 °C. During the addition the reaction mixture turns yellow. After the addition, the ice-bath is removed and the solution is stirred for 20 min (Note 15). The stir bar is removed and the solution is concentrated by rotary evaporation (70 mmHg, bath temperature 40 °C) to afford crude 3 (12.4 g). Vacuum distillation provides (chloromethoxy)(1,1-dimethylethyl)dimethylsilane (3) as a slightly yellow liquid (8.41-8.66 g, 80-82 % yield) (Notes 16 and 17).
D. 4-Methylbenzenesulfonothioic acid, sodium salt (4). Sodium p-toluenesulfinate monohydrate (Note 18) (25.4 g, 0.13 mol. 1.0 equiv), sulfur (4.54 g, 0.14 mol. 1.09 equiv), ethanol (100 mL) and water (100 mL) are added to a 500-mL round-bottomed flask equipped with a PTFE-coated magnetic stir bar (3 x 1 cm). The flask is lowered into a heating mantle and fitted with a Liebig condenser. The mixture is stirred and warmed to reflux for 8 h (Note 19). After the reaction mixture is cooled to room temperature, the residual sulfur is removed by filtration through a 60-mL medium porosity sintered glass funnel into a 1-L round bottom flask, using water (2 x 20 mL) to wash the reaction flask and the collected solid. The filtrate is concentrated by rotary evaporation (50 °C water bath, 70 mmHg) to wet solids (49 g). The flask is equipped with a PTFE stir bar (3 x 1 cm) and water (40 mL) is added. The slightly hazy mixture is stirred at room temperature for 3 h. The solution, which contains some suspended particles, is filtered into a 500-mL round bottomed flask through pad of pre-wetted Celite (3 g) (Note 20) in a 40-mL medium porosity sintered glass funnel, the flask and Celite pad being rinsed with water (4 x 10 mL). The clear filtrate is concentrated by rotary evaporation (50 °C water bath, 70 mmHg) to provide a wet solid (32 g) (Note 21). The flask is equipped with a PTFE stir bar (3 x 1 cm) and a Liebig condenser. Absolute ethanol (80 mL) is added and the heterogeneous mixture is stirred and warmed to reflux over a 30 min period using a heating mantle. Once the mixture reaches reflux, the heating mantle is removed and the mixture is cooled in air to room temperature over the course of 1 h, then is stirred at room temperature for 3 h. The resulting white solid is collected on a 150-mL medium porosity sintered glass funnel, portions of the filtrate being used to rinse all solids out of the flask. The filter cake is washed with absolute ethanol (25 mL), air-dried in the funnel by continued suction (ca 1 h) and then dried for 9 h in a vacuum oven (70 mmHg, 50 °C) to afford 4-methylbenzenesulfonothioic acid, sodium salt (22.4-23.4 g, 82-86 %) (Note 22).
E. 4-Methylbenzenesulfonothioic acid, S-[[[(1,1-dimethylethyl)-dimethylsilyl]oxy]methyl] ester (5). An oven-dried 250-mL round-bottomed flask equipped with a PTFE-coated magnetic stirring bar (3 x 1 cm) is capped with a rubber septum pierced with a nitrogen inlet needle connected to a gas bubbler. A thermocouple thermometer probe is also inserted through the septum (Note 1). The septum is removed momentarily and 4-methylbenzenesulfonothioic acid, sodium salt (4) (5.97 g, 28.4 mmol, 1.03 equiv) and acetonitrile (35 mL) (Note 23) are added and the suspension is stirred. (Chloromethoxy)(1,1-dimethylethyl)dimethylsilane (3) (4.96 g, 27.4 mmol, 1.00 equiv) is added via a 10-mL syringe over 1 min (Note 24). The mixture is stirred vigorously for 4 h (Note 25), then the septum is replaced with a 200-mL addition funnel and t-butyl methyl ether (140 mL) is added dropwise to the stirred mixture over 15 min. The mixture is filtered through a tightly packed pad of Celite (4 cm in diameter x 2.5 cm in height) (Note 26) in a 40-mL sintered glass funnel into a pre-weighed 500-mL round-bottomed flask, using MTBE (3 x 10 mL) as a rinse of the flask and filter cake. The filtrate is concentrated by rotary evaporation (40 °C bath, 70 mmHg) and dried for 3 h at ambient temperature (70 mmHg) to give 4-methylbenzenesulfonothioic acid, S-[[[(1,1-dimethyl-ethyl)dimethylsilyl]-oxy]methyl] ester (5) as a colorless, viscous oil (9.06 g, 99 %) (Notes 27, 28, and 29).
2. Notes
1.
The internal temperature was monitored using a J-Kem Gemini digital thermometer with a Teflon-coated T-Type thermocouple probe (12-inch length, 1/8 inch outer diameter, temperature range -200 to +250 °C). The submitters did not monitor the internal temperature in any of the procedure's steps A-E.
2.
The following reagents and solvents were obtained from Sigma-Aldrich and used as received for Step A: paraformaldehyde (powder, 95%), ethanethiol (97%), sodium methoxide (25% wt % solution in MeOH).
3.
Paraformaldehyde is added in slight excess with the intention to consume all ethanethiol during the reaction to minimize odors.
4.
Ethanethiol was transferred to the flask via a 10 mL graduated glass pipette (2 transfers). After transfer, the pipette was immediately rinsed with bleach.
5.
If a portion of the mixture has splashed onto the walls of the flask, the material should be rinsed down by gentle swirling.
6.
(Ethylthio)methanol (
1) has the following physical and spectroscopic properties: FTIR (microscope) υ (cm
-1): 3395 br, 2968, 2928, 2873, 1453 cm
-1;
1H NMR
pdf (CDCl
3, 500 MHz) δ: 1.32 (t,
J = 7.4 Hz, 3 H), 1.82 (br s, 1 H), 2.73 (q,
J = 7.4 Hz, 2 H), 4.74 (s, 2 H);
13C NMR
pdf (CDCl
3, 125 MHz) δ: 15.2, 24.8, 65.8. Low resolution EI
m/
z calcd for C
3H
8OS 92, found 92. The submitters determined a purity of 91% by GC/MS (Agilent Technologies 7890 GC with 5975C mass spectrometer; column ZEBRON ZB-5, length 30 m, ID 0.25 mm; film thickness 0.25 μm; initial temperature 35 °C for 2 min, final temperature 290 °C for 2 min; rate of temperature increase 10 °C/min; helium gas; flow 0.6636 mL/min; inlet temperature 200 °C; 50:1 split injection); and 83% by NMR. The checker determined a purity of approximately 85% by
1H NMR.
7.
To minimize odors, it is recommended that the material produced in step A be used directly in Step B using the same flask.
8.
The following reagents and solvents were used as received for Step B: dichloromethane (Fisher ACS certified, stabilized), chloro(1,1-dimethylethyl)dimethylsilane (Oakwood Products, Inc., West Columbia, SC), triethylamine (Sigma-Aldrich, 99.5% distilled), hexanes (Fisher, ACS reagent, >98.5%).
9.
The temperature rises to 10 °C after the addition.
10.
The reaction was monitored by
1H NMR. A 0.1 mL aliquot of the reaction mixture was quenched into 1 mL CDCl
3/1 mL sat. aq. NH
4Cl. The layers were separated, then the CDCl
3 layer was concentrated to dryness. The concentrated sample was diluted with CDCl
3 for
1H NMR analysis. Diagnostic peaks: product
2, δ 4.82 (s, 2H, C
H2); starting material
1, δ 4.74 (s, 2H, C
H2). Approx 1.5 % unreacted
1 remained at the 3 h sampling point.
11.
The checker carried out the distillation in a 100-mL pear-shaped flask equipped with a 1-cm oval PTFE stir bar using a 3-cm Vigreaux column at a pressure of 70 mmHg. Three fractions were collected: fr 1, 80 - 120 °C (2.2 g); fr 2, 125-128 °C (28.6 g); and fr 3, 128-134 °C (3.2g). The pot residue was 1.7 g. By GC analysis (conditions in Note
12), fraction 2 was >98 % pure and fraction 3 was 85 - 90% pure. The yield was based on fraction 2. The submitters carried out the distillation at 4.8 mmHg, 58.5 - 61 °C.
12.
(1,1-Dimethylethyl)[(ethylthio)methoxy]dimethylsilane (2) has the following physical and spectroscopic properties: FTIR (microscope) υ (cm
-1): 2957, 2930, 2897, 2858, 1472, 1463 cm
-1;
1H NMR
pdf (CDCl
3, 500 MHz) δ: 0.13 (s, 6 H), 0.91 (s, 9 H), 1.30 (t,
J = 7.4 Hz, 3 H), 2.68 (q,
J = 7.4 Hz, 2 H), 4.82 (s, 2 H);
13C NMR
pdf (CDCl
3, 125 MHz) δ: -4.8, 15.1, 18.4, 24.8, 26.0, 66.2. GC-MS
m/z (relative intensity), 149 (92%, M
+ -
t-Bu), 119 (100%, M
+ -
t-BuMe
2), 91(40%), 89 (61%), 75 (43%), 73 (63%). Purity = 98 % by GC (t
R = 7.5 min; conditions: Agilent DB35MS column; 30 m x 0.25 mm; initial temp 60 °C, ramp at 20 °C/min to 280 °C, hold 15 min).
13.
The submitters report the compound is stable at room temperature for at least 1 month when kept in a stoppered flask.
14.
The following reagents and solvents were used as received for Step C: dichloromethane (Fisher ACS certified, stabilized,), sulfuryl chloride (Acros, 97%).
15.
The temperature rose to 12 °C.
16.
The distillation was carried out in a 50-mL pear-shaped flask containing a PTFE-coated oval magnetic stir bar (1 cm) using a 3-cm Vigreaux column at a pressure of 70 mmHg. Three fractions were collected: fr 1, 30-83 °C (1.18 g); fr 2, 83-87 °C (6.89 g); fr 3, 87-88 °C (1.52 g). GC purity
(Note 12) was 96.5% for fraction 2 and 94.5% for fraction 3. Fractions 2 and 3 were combined. The submitters reported distillation at 70.0-72.5 °C (26 mmHg).
17.
(Chloromethoxy)(1,1-dimethylethyl)dimethylsilane (3) has the following physical and spectroscopic properties: FTIR (neat film, microscope) υ (cm
-1): 2958, 2932, 2901, 2860, 1473, 1464 cm
-1;
1H NMR
pdf (CDCl
3, 500 MHz) δ: 0.21 (s, 6 H), 0.92 (s, 9 H), 5.61 (s, 2 H);
13C NMR
pdf (CDCl
3, 125 MHz) δ: -5.0, 18.0, 25.7, 76.6. GC-MS
m/z (relative intensity), 125 (9 %, M
+ -
t-Bu) 123 (24%, M
+ -
t-Bu), 95 (35 %, M
+ -
t-BuMe
2), 93 (100 %, M
+ -
t-BuMe
2), 73 (25%), 57 (45%). Purity = 96 % by GC (t
R = 5.5 min, conditions in Note
12). The checkers noted the compound decomposes at a rate of 15% per week at room temperature when stored in a glass flask with a glass stopper based on
1H NMR analysis. In the refrigerator, a small amount of decomposition (2-3%) occurred over a two-week period. When stored in the freezer, no decomposition occurred over a two-week period.
18.
The following reagents and solvents were used as received for Step D:
p-toluenesulfinate hydrate (Acros, 98%), sulfur (Fisher, sublimed powder, ethanol (Sigma-Aldrich, 99.5%), Celite (Sigma-Aldrich, acid-washed).
19.
Reaction progress was monitored by
1H NMR as follows. A 0.1 mL reaction aliquot was evaporated to dryness then dissolved in DMSO-d
6. Diagnostic resonances were δ 7.14-7.15 (m, 2 H), 7.61-7.63 (m, 2 H) for product
4 and 7.35-7.37 (m, 2H) for
p-TsSO
2Na. Approximately 1%
p-TsSO
2Na remained at the 7 h timepoint.
20.
The Celite was pre-wetted by filtering 20-mL water through the Celite cake. The hazy filtrate was discarded and the receiving flask rinsed with water prior to filtering the reaction mixture.
21.
The crude material contains 3% sodium
p-toluenesulfinate based on
1H NMR analysis.
22.
4-Methylbenzenesulfonothioic acid, sodium salt (
4) has the following physical and spectroscopic properties: IR (KBr) υ (cm
-1): 3039, 3023, 2981, 2920, 2861, 1934, 1664, 1596, 1494, 1398 cm
-1;
1H NMR
pdf (500 MHz, DMSO-d
6) δ: 2.30 (s, 3 H), 7.14-7.15 (m, 2 H), 7.61-7.63 (m, 2 H);
13C NMR
pdf (125 MHz, DMSO-d
6) δ: 20.7, 124.0, 128.0, 138.2, 152.6; exact mass (electrospray)
m/
z calcd for C
7H
7O
2S
2 (M-Na) 186.9893, found 186.9895. The material contained 0.2 wt% water based on Karl Fischer titration. Weight percent purities of 94% and 97% (2 runs) were determined based on quantitative
1H NMR analysis (DMSO-d
6) using dichloroethane as internal standard, based on 2 weighings (samples 30-60 mg each), 4 acquisitions using a pulse delay of 10 seconds, and 8 independent peak integrations. The material with 94 wt% purity was purified by slurrying as follows: Compound
4 (22.6 g) and 95% ethanol (80 mL) were added to a 500-mL round bottomed flask equipped with a PTFE oval-stir bar (3 x 1 cm). The stirred mixture was heated to reflux (remains a slurry) with a heating mantle, then cooled in air with stirring to ambient temperature over 1 h and stirred for an additional 3 h. The material was filtered into a 60-mL sintered glass funnel, washed with absolute ethanol (25 mL), and dried under vacuum (70 mmHg, 50 °C) to afford product (17.9 g, 79% recovery). NMR quantitative assay indicated 98 wt% purity.
23.
The following reagents and solvents were used as received for Step E: acetonitrile (Fisher Optima, water content 0.001%),
t-butyl methyl ether (>98.5%, Sigma-Aldrich), Celite (Sigma-Aldrich, acid-washed).
24.
The reaction warms to 27 °C over 10 min, then cools slowly to room temperature over 30 min.
25.
The reaction remains heterogeneous throughout. The reaction was monitored by
1H NMR as follows. A 0.1 mL reaction aliquot was added to 1 mL MTBE, filtered, and concentrated to dryness. The sample was dissolved in CDCl
3 for analysis. Diagnostic peaks were δ 5.61 (s, 2H) for unreacted
3 and 5.41 (s, 2H) for product
5. Less than 1% starting material remained at the 3 h timepoint.
26.
The Celite was pre-wetted by filtering 20-mL MTBE through the Celite cake. The filtrate was discarded and the receiving flask rinsed with MTBE prior to filtering the reaction mixture. The submitters used diethyl ether in this experiment instead of MTBE.
27.
4-Methylbenzenesulfonothioic acid, S-[[[(1,1-dimethylethyl)-dimethylsilyl]oxy]methyl] ester (
5) has the following physical and spectroscopic properties: mp 33-34 °C (MTBE/hexanes); FTIR (dichloromethane cast film) υ (cm
-1): 2955, 2930, 2886, 2858, 1595, 1493, 1472, 1464, 1444 cm
-1;
1H NMR
pdf (CDCl
3, 400 MHz) δ: -0.03 (s, 6 H); 0.76 (s, 9 H), 2.42 (s, 3 H), 5.41 (s, 2 H), 7.27-7.32 (m, 2 H), 7.84-7.86 (m, 2 H);
13C NMR
pdf (100 MHz, CDCl
3) δ: -5.3, 18.1, 21.8, 25.6, 71.5, 127.3, 129.8, 144.0, 144.6; exact mass (electrospray)
m/
z calcd for C
14H
24NaO
3S
2Si 355.0828, found 355.0824; GC-MS
m/z (relative intensity), 275 (6%, M
+ -
t-Bu), 245 (59%, M
+ -
t-BuMe
2), 91 (39%), 75 (100%), 73 (30%). Anal. calcd. for C
14H
24O
3S
2Si: C, 50.56; H, 7.27; S, 19.28; found C, 50.44; H, 6.83; S, 19.61. Weight percent purities of 87% and 90% (2 runs) were determined based on quantitative
1H NMR analysis (CDCl
3) using dichloroethane as internal standard, as outlined in note 22.
28.
A purified sample of
5 was obtained by crystallization as follows: Compound
5 (7.1 g) was dissolved in MTBE (10 mL). Residual solids were removed by filtration through a 0.45 μm PTFE syringe filter (25 mm, Millipore catalog # SLCR025NS) into a 100-mL round-bottomed flask. Hexanes (20 mL) were added and the solution was held in a -20 °C freezer for 20 h. (Obtaining crystals for the first time at small scale required 6 days in the freezer; thereafter, crystallization typically initiated within a few hours). The product was isolated by filtration on a 40-mL sintered glass funnel, using MTBE/hexanes (1:1) (5 mL, -20 °C) as a wash, to provide colorless cubic crystals (4.1 g 59% recovery). Weight percent purities of 97% and 98 % (2 runs) were determined based on quantitative
1H NMR analysis (CDCl
3) using dichloroethane as internal standard, as outlined in note 22.
29.
The submitters report the compound decomposes on silica gel and on neutral Grade III alumina. The compound has been kept without change (
1H NMR) at room temperature for 1 week and is stable in the freezer for at least 2 months.
Handling and Disposal of Hazardous Chemicals
The procedures in this article are intended for use only by persons with prior training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011 www.nap.edu). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
These procedures must be conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
3. Discussion
During the course of synthetic studies directed towards the antitumor agent MPC1001,
2 a need arose for a protected bivalent sulfur unit so constituted that it could be introduced by reaction with a carbanion and later dismantled to release a sulfhydryl group under mild conditions. Although numerous sulfur protecting groups are known,
3 our precise requirements prompted us to investigate the CH
2OSiMe
2Bu-
t group for sulfur protection and the reagent 4-MeC
6H
4SO
2-SCH
2OSiMe
2Bu-
t (
5) for introducing sulfur protected in this manner.
4 Other sulfonothioic acid esters such as PhSO
2-SPh have been used for sulfenylation of carbanions,
5 and the use of a silyl group offered the possibility of controlling the deprotection conditions by changing the substituents on silicon.
6 Although chloromethoxysilanes have been used for protection of alcohols,
7 they do not appear to have been used for sulfur protection.
A direct method for preparing reagent (
5) appeared to be by way of reaction E above, and so we prepared the two components, salt (
4) and the chloromethoxysilane (
3). Both were made by the literature methods
8,9 which are described in detail here. Reaction of the salt with silane (
3) afforded the desired sulfenylating agent (
5).
In order to test if the CH
2OSiMe
2Bu-
t group could indeed serve as an effective form of protection for bivalent sulfur, we treated a number of simple thiols with reagent (
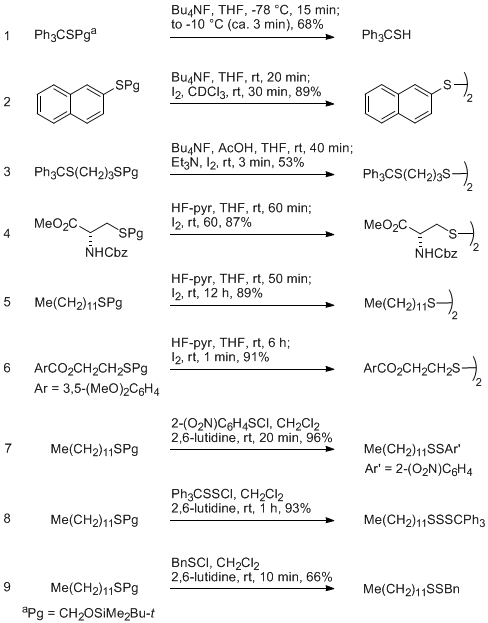
3) so as to generate the sulfides shown in Table 1.
4 DMF is a satisfactory solvent and proton sponge or 2,6-lutidine gave the best results, depending on the particular case.
Table 1. Protection of thiols
Several methods for deprotection of the sulfides were examined (Table 2).
4 Treatment with Bu
4NF in THF; Bu
4NF and AcOH in THF, or HF.pyridine in THF were all suitable, and it was usually convenient to oxidize the thiols in situ by addition of iodine. In a few cases the protecting group was removed by reaction with a sulfenyl halide (2-nitro-benzenesulfenyl chloride, α,α-diphenylbenzenemethanesulfenyl chloride, benzenemethanesulfenyl chloride).
Table 2. Deprotection
The stability of the protecting group was evaluated
4 by exposing
n-C
12H
25SCH
2OSiMe
2Bu-
t (
6) to a variety of conditions. The compound is stable to H
2/Pd/C in methanol-dichloromethane and to H
2/Rh/Al
2O
3/ethyl acetate. An
O-triethylsilyl ether can be selectively deprotected in the presence of (
6), using H
2/Pd/C in methanol-dichloromethane. The protecting group appears to survive typical conditions for removal of a Troc group (zinc dust in acetic acid-diethyl ether) and an Fmoc group can be removed in its presence by using piperidine. Hydride reducing agents either have no effect (sodium borohydride) or little effect (lithium aluminum hydride, diisobutylaluminum hydride). (PhS)
2CH
2 can be deprotonated with butyllithium with very little decomposition (4%) of the test substrate. Acidic reagents (trifluoroacetic acid,
p-toluenesulfonic acid hydrate, pyridinium
p-toluenesulfonate-MeOH, boron trifluoride etherate) are not compatible with the protecting group, except for pyridinium
p-toluenesulfonate in dichloromethane and exposure to silica gel during chromatography. A primary alcohol can be converted into the corresponding bromide (tetrabromomethane, triphenylphosphine) without affecting the protecting group, but oxidizing agents (pyridinium chlorochromate, Dess-Martin periodinane, 2-iodoxybenzoic acid (IBX), Swern conditions) are not compatible with the protecting group. A primary alcohol can be silylated with triethylsilyl triflate in the presence of (
6).
The sulfenylating agent (
5) has been used to introduce the protected sulfur, as shown in eq 1.
4 Sulfur deprotection is illustrated in eq 2,
10 which represents the result of a single experiment. A more sophisticated use of the sulfenylating agent (
5) as well as subsequent deprotection in a synthetically complex setting is summarized in Scheme 1.
10Scheme 1. Sulfenylation with reagent 5 and deprotection
Appendix
Chemical Abstracts Nomenclature (Collective Index Number);
(Registry Number)
(Chloromethoxy)(1,1-dimethylethyl)dimethylsilane; (119451-80-8)
Chloro(1,1-dimethylethyl)dimethylsilane; (18162-48-6)
4-(Dimethylamino)pyridine; (1122-58-3)
(1,1-Dimethylethyl)[(ethylthio)methoxy]dimethylsilane; (119451-79-5)
Ethanethiol; (75-08-1)
(Ethylthio)methanol; (15909-30-5)
4-Methylbenzenesulfonothioic acid, S-[[[(1,1-dimethylethyl)dimethylsilyl]-oxy]methyl] ester; (1277170-42-9)
4-Methylbenzenesulfonothioic acid, sodium salt (1:1); (3753-27-3)
Paraformaldehyde; (30525-89-4)
Sodium methoxide; (124-41-4)
Sodium p-toluenesulfinate hydrate (TolSO2Na·H2O); (207801-20-5)
Sulfur; (7704-34-9)
Sulfuryl chloride; (7791-21-5)
Triethylamine; (121-44-8
)
 |
Derrick Clive was born in London and was educated at Imperial College where he obtained a B.Sc. (Special) in Chemistry, and then a Ph.D. in Professor Barton's group. Dr. Jack E. Baldwin (now Sir Jack) assisted in the supervision of these postgraduate studies. Derrick then held a postdoctoral position at Harvard in R. B. Woodward's group. In 1975 he joined the Chemistry Department of the University of Alberta, where he is now Professor of Chemistry. He has published over 200 papers on the development of general synthetic methods - involving mainly selenium chemistry and radical cyclization - and on the total synthesis of complex natural products with significant biological properties. |
 |
Lihong Wang was born in Zhoushan, Zhejiang Province, and obtained his B.Sc. at Fudan University. He stayed at Fudan University to begin his graduate studies, but moved to the University of Alberta in 2006 for his Ph.D. under the supervision of Professor Clive. Lihong's research has been supported by a number of Scholarships and is in the area of synthetic methodology and natural product synthesis. After obtaining his Ph.D. in 2011 he joined Professor Nicolaou's group at the Scripps Institute as a postdoctoral fellow. |
Copyright © 1921-, Organic Syntheses, Inc. All Rights Reserved